
小儿β地中海贫血简介:β地中海贫血(β-mediterranean anemia)是指β链的合成受部分或完全抑制的一组血红蛋白病。
- 发病部位
- 传染性
- 治愈率
- 多发人群
- 相关症状
- 并发疾病
- 小儿β-珠蛋白生成障碍贫血
- 血液血管
- 无传染性
- 10%
- 儿童
- 鼻梁低,肝功能衰竭,食欲不振
- 黄疸,贫血,肺炎
- 挂号科室
- 治疗方法
- 治疗周期
- 治疗费用
- 临床检查
- 无
- 儿科,血液科
- 药物治疗
- 1-3个月
- 市三甲医院约(3000-8000元)
- 红细胞渗透脆性试验,红细胞检查,骨髓象分析
- 常见药品
- 羟基脲片,重组人红细胞生成素-β注射液,地中海贫血筛查血样标本采集卡
- 在线购药
疾病症状:
一、症状
根据严重程度又可分为极轻型、轻型、中间型及重型四等级。极轻型地中海贫血无贫血症状因此又叫做地贫基因携带者,轻型地中海贫血仅有轻度贫血,多在体检时才发现,这两型患者一般都不知自己带有这种遗传基因。中间型患者中度贫血,可接受药物治疗和脾切除手术,提高血红蛋白,但仅仅是改善贫血,不能治愈,相对较重的还要输血。重型地中海贫血是一种非常严重的疾病,出生后3-6个月发病,他们需要终生定期的输血和接受除铁剂治疗。各类地中海贫血之间又可互相组合,可与各种异常Hb组合(如HbE/β地中海贫血),这一组疾病又称地中海贫血综合征,均属常染色体不完全显性遗传。
二、诊断
1.轻型β地贫
只有轻到中度贫血而无症状。感染和妊娠时贫血加重。HbF正常或轻度增加(不超过5%)。寻查遗传基因,父或母为β海盆杂合子。除外其他珠蛋白生成障碍性贫血和缺铁性贫血。凡符合上述条件可诊断本病本型。
2.中间型β海贫
症状和体征介于重型和轻型β海贫之间。贫血程度、肝脾肿大、面部改变与骨骼改变均较重型为轻,发病较迟,病程较良性。本病如不治疗,多于5岁前死亡。多种不同基因引起的中间型珠蛋白生成障碍性贫血,需依据基因分析和Hb结构分析进行诊断。
3. 重型β海贫
有严重贫血并出现症状。临床多见患儿出生时无症状,生后3~6 个月内发病者占50%,偶有新生儿期发病者。发病年龄愈早,病情愈重。较早出现的严重溶血性贫血的相应临床表现,如进行性贫血,黄疸,伴骨骼改变,首先发生于掌骨,再至长骨、肋骨,最后为颅骨,形成特殊面容(Downs面容):头大、额部突起、两颧略高、鼻梁低陷,眼距增宽,眼睑水肿。皮肤斑状色素沉着。食欲不振,生长发育停滞,肝脾日渐肿大,以脾大明显,可达盆腔。X线可见骨质稀疏、骨皮质变薄、髓腔增宽、外板骨小梁条纹清晰呈直 立的毛发样等,可有病理性骨折。患儿常并发支气管炎或肺炎。并发含铁血黄素沉着症时因过多的铁沉着于心肌和其他脏器如肝、胰腺等而引起该脏器损害的相应症状,其中最严重的是心力衰竭和肝纤维化及肝功能衰竭,是导致患儿死亡的重要原因之一。凡临床符合有重度溶血性贫血,HbF>30%者,可初步诊断重型β海贫。为进一步确 定诊断可做α和β珠蛋白链的合成比率测定和基因分析。家系调查可证明患者的父母均为轻型β海贫。并能除外HbF增加的其他珠蛋白生成障碍性贫血,如HbF持存,F(δβ)、A~2F和βγ珠蛋白生成障碍性贫血。严重的慢性进行性贫血,需依靠输血维持生命,3~4 周输血1 次,随年龄增长日益明显。
根据临床表现和血液检查特别是HbF含量增高及家系调查可确诊,有条件者可进一步作肽链分析或基因诊断。
疾病病因:
一、发病原因
正常成人血红蛋白中的珠蛋白,是由四条肽链所组成的,本病是由于珠蛋白基因的缺失或点突变所致。组成珠蛋白的肽链有4种,即α、β、γ、δ链,分别由其相应的基因编码,这些基因的缺失或点突变可造成各种肽链的合成障碍,致使血红蛋白的组分改变。通常将地中海贫血分为α、β、γ和δ等4种类型,其中以β和α地中海贫血较为常见。β珠蛋白基因位于11号染色体短臂1区2带(简记:11P12),本病除少数几种为几个核苷酸缺失外,绝大部分都是该基因点突变(单个核苷酸置换、增加或缺失)所致。全世界已发现100种基因突变类型,我国有20种。染色体上的2个等位基因突变点相同者称纯合子;等位基因的突变不同者称“双重杂合子”;同源的染色体上只有1个突变者称“杂合子”。非典型β地贫杂合子:指位于β珠蛋白基因远侧的启动区域位点控制区(locus control region,LCR),这些并非在β链上的分子缺陷(非典型β地贫基因)与典型的β地贫基因组合形成双重杂合子。突变致β链合成部分受抑制者称“β 地中海贫血”;致β链完全受抑制者称“βo地中海贫血”。肽链合成的抑制涉及δ链,称为β 海洋性贫血。
根据β基因缺陷所产生的杂合子和纯合子的不同,其临床表现亦有差异,按照病情轻重可分轻型和中间型、重型三种类型。
1.轻型β-地中海贫血
(β-mediterranean anemia minor或trait)是β0、β 或δβ基因,包括双重突变杂合体(指病变的β珠蛋白基因的核苷酸序列上同时有2种突变点)的杂合子状态,主要表现为轻度小细胞、低色素贫血。尤其婴儿期明显,可有轻度黄疸和脾大。
2.中间型β地中海贫血
即中间型β海洋性贫血(β-mediterranean anemia intermedia),包括β 基因纯合子、某些β0或β 双重杂合子,非典型β地贫杂合子、合并α地贫血和δβ地贫。其临床表现介于重型与轻型之间,特点:发病年龄较晚(常于4~5岁),黄疸可有可无,对外界感染抵抗力弱,骨骼改变较轻。轻度或中度肝脾肿大。中度贫血,Hb 60~90g/L,输血量小或不必输血仍可维持生命;外周血涂片红细胞形态与重型类似。红细胞渗透脆性减低,HbF含量增高,HbA2可稍高、正常或降低。家系调查证实为上述基因传递。
3.重型β地中海贫血
(β-mediterranean anemia major)又称库理贫血(Cooleys anemia),患儿在临床上呈慢性溶血性贫血。贫血和缺氧刺激红细胞生成素(EPO)的合成进而刺激髓外造血。这是β地中海贫血纯合子、β0与β 地中海贫血双重杂合子或δβ0纯合子因β链生成完全或几乎完全受到抑制,以致含有β链的HbA合成减少或消失,而多余的α链则与γ链结合而成HbF(α2γ2),使HbF明显增加,由于HbF的氧亲和力高,致患者组织缺氧。过剩的α链沉积于幼红细胞和红细胞中,形成α链包涵体附着于红细胞膜上而使其变僵硬,在骨髓内大多被破坏而导致“无效造血”;部分含有包涵体的红细胞虽能成熟并被释放至外周血,但当它们通过微循环时就容易被破坏;这种包涵体还影响红细胞膜的通透性从而导致红细胞的寿命缩短。贫血引起的骨髓扩张,导致头骨和面部的变形,产生特征性地中海贫血外貌。贫血使肠道对铁的吸收增加,加上在治疗过程中的反复输血,使铁在组织中大量贮存,导致含铁血黄素沉症。是我国常见的一种地中海贫血。本症患者双亲均为β-地贫杂合子,其子女中获得重症β-地贫几率为25%,50%为杂合子,余25%为正常。
二、发病机制
正常成人的血红蛋白有三种:HbA(a.β)占血红蛋白总量的95%~97%,由2条a链与2条β链组成;HbAz(a。,β) 链与2条β链组成; 占规~3% ;HbF(a,δ)<2%,由2条a链与2条δ链组成。HbF是胚胎期的主要血红蛋白,妊娠后34~36周以前HbF占血红蛋白总量的90%~95%,新生儿占50%~85%,一岁左右降至成人水平。HbAz在胚胎中出现较晚,出生6~12个月后与成人含量相同。HbA则出生后增多很快,至6月后达成人水平。而胚胎中最早出现的是Hb Gowerl(E。。。)、Hb Gower巨(a。。。)及Hb Portland(8。y;),这些Hb在胚胎10周后逐渐被HbF所替代,正常成人Hb 中Gowerl已经消失,在脐血中可有极微量的Hb Portlqnd。
β-地贫的许多病理生理和临床表现均与珠蛋白链合成不平衡有关。地中海贫血是由于珠蛋白基因组织和结构的多种突变,使基因表达发生了部分β少,或完全;a、β在肝肝内制造障碍(平常用a、β-或a、β 表示),导致一种或几种正常的珠蛋白链合成减少或缺如所造成的组成珠蛋白的肽链高度异质性综合征。由于β链的合成受到抑制,故HbA(α2β2)的合成可减少或不存在,在杂合子,多余的α链与代偿性增多的δ链结合,使AbA2(α2δ2)增加。在纯合子由于β链的显著减少,α链相对增加,多余的α链与γ链结合,故HbF(α2γ2)成为红细胞中主要的Hb成分。由于HbF较HbA的氧亲合力高,在组织中不易释出氧,故患者常有组织缺氧,缺氧引起红细胞生成素的大量分泌,刺激骨髓造血功能,红骨髓极度扩张,因而引起一系列骨骼改变。由于α与β链之间的不平衡,过剩的α链可聚合成极不稳定的α2、α3,或α4,易变性沉积于幼红细胞和红细胞中而形成α链包涵体,由于包涵体附着于红细胞膜而使红细胞膜变僵硬,易受机械性损伤,在骨髓内破坏而不能全部进入血液循环,导致“无效造血”。部分含有包涵体的红细胞虽然成熟并释放至外周血,但这些红细胞通过微循环时,易被破坏,使其寿命缩短,此外,红细胞的包涵体还影响红细胞的通透性,进一步使其寿命缩短,导致溶血性贫血。
HS的遗传缺陷有异质性,大多数属常染色体显性遗传(占75%),少数为常染色体隐性遗传(占25%)。一般有明显家族史,但10%-20%的病人可无明显家族史,其病因可能为基因的自我突变,也可能与变异的表现型或常染色体遗传有关,随着分子生物技术的发展,目前已基本阐明HS的基因及膜蛋白。
贫血促进肠道对铁的吸收增加(达80%),加以铁利用障碍和治疗过程中的反复输血。使心、肝、脾、骨髓及皮肤等组织沉积大量的铁,导致含铁血黄素沉着症,晚期产生继发性血色病,心肌和肝功能等遭受损害,糖尿病和其他内分泌障碍等。不同类型海贫的临床表现差别极大,最重者可于胎儿出生前即死亡,最轻者可以终身不贫血,无症状。临床所见大多是介于这两者之间的贫血患者,具有低色素小红细胞和靶形红细胞,并有血红蛋白成分的各种改变。重型多生长发育不良,常在成年前死亡。轻型及中间型患者,一般可活至成年并能参加劳动,倘注意节劳及饮食起居,可以减少并发症、改善症状。
疾病预防:
小儿β地中海贫血预防
预防在本病中起重要作用。婚前地中海贫血筛查,避免轻型地中海贫血患者联婚,可明显降低重型/中间型地贫患者出生的机会。推广产前诊断技术,对父母双方或一方地贫基因携带者,孕4个月时,采集胎儿绒毛、羊水细胞或脐血,获得基因组DNA以聚合酶链反应(PCR)技术对高危胎儿进行产前诊断,重型/中间型患儿应终止妊娠。
疾病鉴别:
小儿β地中海贫血鉴别
本病需与以下疾病鉴别:
1.缺铁性贫血
需注意与发生于婴儿期重症缺铁性贫血区别。轻型地中海贫血的临床表现和红细胞的形态改变与缺铁性贫血有相似之处,故易被误诊。但缺铁性贫血是由于体内缺少铁质而影响血红蛋白合成所引起的一种常见贫血。在红细胞的产生受到限制之前,体内的铁贮存已耗尽,此时称为缺铁。这种贫血特点是骨髓、肝、脾及其他组织中缺乏可染色铁,血清铁浓度和血清转铁蛋白饱和度均降低。世界各地可见。其病因:一是铁的需要量增加而摄入不足,二是铁的吸收不良,三是失血过多等,均会影响血红蛋白和红细胞生存而发生贫血。鉴别:缺铁诱因,血清铁蛋白含量减低,骨髓外铁粒幼红细胞减少,红细胞游离原叶琳升高,铁剂治疗有效等。
2.血红蛋白E病
该病是由于珠蛋白基因发生点突变,β-珠蛋白链上某种氨基酸被另一种氨基酸取代,使血红蛋白的性质和功能发生变化而引起的疾病。与本病相似,但本病Hb电泳可见HbE>30%。
3.红细胞G-6PD缺乏所致先天性非球形细胞性溶血性贫血(CNSHA)
重型者与本病临床表现相似。但前者自幼年起出现慢性溶血性贫血,表现为贫血、黄疸、脾肿大;可因感染或服药而诱发急性溶血。G-6PD活性降低,红细胞Heinz小体生成试验阳性,HbF含量正常可鉴别。
4. 传染性肝炎或肝硬化
因HbH病贫血较轻,还伴有肝脾肿大、黄疽,少数病例还可有肝功能损害,故易被误诊为黄疽型肝炎或肝硬化。但通过病史询问、家族调查以及红细胞形态观察、血红蛋白电泳检查即可鉴别。
5.新生儿ABO溶血病
指母婴血型不合引起的同族免疫性溶血,仅见于胎儿新生儿期,可出现暂时球形细胞增多,应与新生儿HS相鉴别。但这种球形红细胞可随血中抗体降低而消失。血清学检查抗A(或抗B)抗体阳性,Coombs试验阳性,无阳性家族史可以鉴别。
疾病检查:
小儿β地中海贫血检查
1、血象
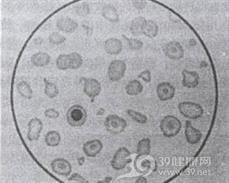
Hb 100~120g/L,红细胞<2.0×1012/L,红细胞大小不等,中心淡染扩大,靶形红细胞多见,呈小细胞低色素性贫血,中央浅染。嗜碱性点彩红细胞、多嗜性红细胞、有核红细胞增加,白细胞及血小板数增加,网织红细胞增高(≤0.1),外周血涂片红细胞异形明显,可见梨形、泪滴状、小球形、三角形、靶形及碎片。并发脾功能亢进。血浆及红细胞内维生素E含量显著下降,与病情呈正相关;超氧阴离子自由基增加。
2.红细胞盐水渗透性试验
红细胞渗透脆性减低,0.3%~0.2%或更低才完全溶血。根据病情的不同程度的而不同。
3.自溶试验与酸化甘油溶解试验
自溶试验室在37℃孵育48小时后自发溶血的程度。正常人<5%,HS时自溶增加到15%-45%,加入葡萄糖或ATP可抑制溶血。由于本试验需无菌技术及仪器,并要48小时后才可获结果,目前已少用。不同红细胞在酸化甘油中溶解速度不同,球形红细胞比正常红细胞的溶解度快10倍以上。本试验敏感性高,阳性率达100%,适用于初筛试验。
4.HbF测定
诊断重型β珠蛋白生成障碍性贫血的重要依据。HbF含量轻度升高(<5%)或明显增高(20%~99.6%);HbA2常降低、正常或中度增高,HbA2 3.5%~8.0%。应注意鉴别,某些再生障碍性贫血、急性白血病,幼年慢性粒细胞性白血病患者的HbH 增高, HbZ ü rich 和 Tocoma 等疾病, HbA2 亦可增高。将 Hb 洗脱用光度计比色(波长413nm的721型分光光度计),计算 HbA2 含量,正常值< 3.5 %。
5.骨髓象
有贫血者呈红系增生性改变,核红细胞增生极度活跃,粒∶红比值倒置,以中、晚幼红细胞为主,胞体小,核固缩,胞浆少偏蓝,甲基紫染色可见晚幼红细胞含包涵体(仅链沉淀),重型β地中海贫血电镜下可查见 a 肽链变性珠蛋白小体。
6.肽链分析
采用高效液相层析分析法可测定β肽链的含量,Cooley贫血时,β/α比值<0.1(正常值为1.0~1.1)。因本病多为点突变,故常用PCR加ASO才能明确突变点,我国各民族的β地贫基因突变情况有一定差异,南方汉族的突变基因以CD41-42(-TCTTT)、CDL7(A →T)、IVS-Ⅱ-654(C →T)和TATA-box28(A →G)为主,占85%~90%。双重杂合子的突变组合可达近100种。而地中海贫血时有不同程度的比例失衡。另外应用 8M 尿素进行肽链裂解,使α链和β链分开,可分别检测各肽链的病变。
7.膜收缩蛋白定量
较电泳更敏感更精确。
8.骨骼X线检查
骨髓腔增宽,皮质变薄和骨质疏松,颅骨的内外板变薄,板障加宽和短发样骨刺形成。
9.常规做X线
B超、心电图等检查。
疾病就诊:
疾病治疗:
小儿β地中海贫血一般治疗
一、治疗
重型β地中海贫血,高量输血联合除铁治疗是基本的治疗措施。造血干细胞移植(包括骨髓、外周血、脐血)是根治本病的惟一临床方法,有条件者应争取尽早行根治手术。轻型地中海贫血不需治疗,注意休息和营养,积极预防感染。适当补充叶酸和维生素E。中间型α地中海贫血应避免感染和使用过氧化性药物,中度贫血伴脾肿大者可做切脾手术,一般不输血,但遇感染,应激,手术等情况下,可适当予浓缩红细胞输注。
1.输浓缩红细胞:目前重要疗法之一
A)低量输血:
仅适用于中间型α和β地贫,不主张用于重型β地贫,单纯的输血或输红细胞最终导致血色病。中等量输血疗法,使血红蛋白维持在60~70g/L。实践证明,这种输血方法虽然使重型患者有望摆脱近期死亡的威胁,但患者的生存质量随年龄增长越来越差。相当一部分患者于第2个十年内因脏器功能衰竭而死亡。
B)高量输血:
以使患儿生长发育接近正常和防止骨骼病变。先反复输浓缩红细胞,使患儿血红蛋白含量达120~140g/L,然后每隔3~4周Hb≤80~90g/L时输注浓缩红细胞10~15ml/kg,使Hb含量维持在100g/L以上。 其优点是纠正机体缺氧;减少肠道吸收铁;抑制脾肿大;纠正患儿生长发育缓慢状态。但本法容易导致含铁血黄素沉着症,故应同时给予铁鳌合剂治疗。
2.脾切除术
可致免疫功能减弱,应在5~6岁以后施行并严格掌握适应证。中度/重度贫血(Hb<80g/L)无黄疸的HbH患病者,行切脾术疗效极佳,可使Hb上升至90~110g/L,若术前Hb>80g/L或慢性溶血性黄疸者,切脾常无效。
对重型β地中海贫血,脾切除一般无效。重型β地贫伴脾功能亢进者,行脾切除能减少对输血的需要量,并减轻体内铁负荷。但切脾面临着发生严重感染等致命并发症的危险,故脾切除应有严格的指征:输血量、 一般年龄应在5岁以上,伴有脾功能亢进(红细胞破坏增加,持续的白细胞或血小板减少)、巨脾引起压迫症状。有学者认为,脾脏正常患者每年每公斤体重的输血商>1,可作为临床判断早期脾功能亢进的指标。 脾大部分栓塞治疗(PSE):由于脾脏为重要的免疫器官,为避免脾切除后继发性免疫功能低下和凶险的感染,有学者提出脾大部分栓塞治疗(PSE)代替脾切除,Politis等对比了PSE与脾切除术后5年的追踪随访资料,结果显示两组病人输血量较前都有减少,但PSE组血中IgM浓度明显高于脾切除组,且感染的发生率也明显低于后者。但有资料显示PSE对中间型β地贫效果较满意,对重型β地贫的疗效不满意,可能与重型β地贫粗大颗粒的红细胞包涵体主要在骨髓破坏有关。以上2种手术后均可导致肝脏代偿性溶血,引起明显肝大、纤维化加重。
3.铁螯合剂
因长期高量输血、骨髓红细胞造血旺盛,以及胃肠道铁吸收的增加,常导致体内铁超负荷易合并血色病,损害心肝、肾及内分泌器官功能。当患者体内的铁累积到20g以上时,则可出现明显的中毒表现,故应予铁螯合剂治疗,通常在规则输注红细胞 1 年或 10~20 单位后进行铁负荷评估,如有铁超负荷(例如 SF >1000μg/L)、则开始应用铁鳌合剂。1岁内使用铁螯合剂,其副作用如骨骼畸形、生长抑制的发生率明显升高,一般主张2~3岁后或患儿接受10~20次输血后并有铁负荷过重的证据,血清铁(SF)>1000μg/L,血清转铁蛋白完全饱和才开始除铁治疗。当前临床广泛使用的是去铁胺(Deferoxamine,DFO), 可以增加铁从尿液和粪便排出,但不能阻止胃肠道对铁的吸收。剂量:20~50mg/(kg·d),加注射用水或生理盐水用便携式输液泵每天(或每晚)腹壁皮下注射8~12h,每周连用5~6天,或加入等渗葡萄糖液中静滴8~12小时;每周5~7天,长期应用。用药前后应作SF、尿铁的监测。若SF>3000μg/L或者有铁负荷继发心脏病时,可予DFO 50~70mg/(kg·d)持续24h静脉滴注。使用铁整合剂时加用维生素C口服可增加尿中铁的排泄量1倍。但维生素 C可将铁从储备部位动员出来并通过氧化代谢间接影响心肌细胞,故在重度铁负荷时不宜使用大剂量维生素 C,一般每天口服100~200mg。在停用DFO期间也不应坚持服维生素 C。长期使用DFO一般无明显的毒副作用,注射局部反应、皮疹、疼痛,无需停药。但铁负荷轻者使用大剂量DFO可出现白内障、听力丧失、长骨生长障碍等,应引起临床重视。
近十年来,国外一系列新型口服铁螯合剂如defefipone(L1)、多价阴离子胺(the poly anionicanine,HBED),多价氮替代物(the substituted poly aza compounox TR coll)、PIH等相继问世,在动物实验中已证实长期服用能有效地降低机体铁负荷,但只有L1试用于人体。 通过高量输血与除铁治疗可维持患者正常生长发育及达到正常人的生活质量及寿命,但必须终身承受沉重的经济负担,可能的输血相关合并症及心理负担。
4.异基因造血干细胞移植(HSCT)
目前能根治重型β地贫的方法,包括骨髓移植(BMT)、脐血移植(UCBT)、外周血造血干细胞移植(PBSCT)和宫内造血干细胞移植(IUSCT)。如有 HLA 相配的造血干细胞供者;应作为治疗重型β地贫的首选方法。迄今,全世界已成功开展HSCT 1200例,其中BMT达1000余例,PBSCT 10例,UCBT约30例,IUSCT 2例。目前IUSCT成功所需单个有核细胞数、移植的最佳胎龄、植入后的状态等尚需进一步深入研究。研究发现,重型地贫的HSCT有其自身的特点。
A)受体的选择:
移植前病者应做危险因素评分:去铁胺应用史(“0”分为规则使用,即经1次规则输血后18个月开始,每周至少5天,皮下输注持续8~10h。“1”分为不规则使用,未达上述任一标准)、肝大(“0”分为肝活检无纤维化。“1”分为纤维化)。将其分为3类:Ⅰ类(0分),Ⅱ类(1~2分),Ⅲ类(3分)。 BMT效果顺序为Ⅰ>Ⅱ>Ⅲ类;无病存活率分别为85%、80%、53%,纤维化及铁负荷是重要危险因素。
B)供体选择:
血缘相关HLA全相合的供体:同胞HLA相合供体BMT可治愈80%的重型β地贫,但仅25%~30%患者可找到HLA相合的家系成员供体。 血缘相关HLA不全相合(包括同胞、双亲)或单倍体相合供体:生存率及无病生存率分别为58%和26%,排斥率41%,总病死率44%。因此,找不到HLA相合供体,且无法接受输血及去铁治疗者,才考虑进行此类移植。 非血缘相关(UD)供体:Dihi报道3例地贫UD-BMT:1例植活,1例回复地贫状态,1例死于移植物抗宿主病(GVHD);Miano报告8例,4例成功,3例排斥,1例死亡。发生排斥是此类移植面临的主要问题。IUSCT近年已有成功报道。1996年Touraine等用胎肝造血干细胞宫内移植治疗2例宫内确诊为地贫胎儿,1例死亡,1例生下无病生存已达4年之久。但Monni等用父亲骨髓CD34细胞经胎儿腹腔注射治疗1例胎龄为10周的重型B地贫胎儿未获成功。
5.基因治疗
从分子水平上纠正致病基因的表达,即基因治疗。应用化学药物可增加 γ基因表达或减少α基因表达,以改善β地贫的状,已用于临床的药物有经基脲、5 -氮杂胞苷(5~AZC)、阿糖胞苷、马利兰、异烟肼等。中间型效果明显,重症者一般用药初期效果明显,随治疗时间延长,效果渐差。人们正对这种药物调控基因的机制深入研究,从而加深对珠蛋白基因表达的遗传调控的认识。
6.其途径有以下两点
A)将正常的β珠蛋白的基因导入患者的造血干细胞;
以纠正β地贫的遗传缺陷。需解决:转移的外源珠蛋白基因能在细胞和整体达到高度表达;必须分离、纯化获得用于基因传导的人类造血干细胞;α基因与β基因之间表达协同一致性。此外,转导的外源性基因必须随珠蛋白基因系统在个体发育过程中适时表达仍处于实验研究阶段。
B)采用某些药物调节珠蛋白基因的表达:
采用某些药物调节珠蛋白基因的表达以平衡α、β珠蛋白的肽链水平。目前临床应用以调节珠蛋白基因表达的药物有白消安(马利兰),羟基脲,丁酸盐,阿糖胞苷(Ara-C),红细胞生成素(Epo)和异烟肼(雷米封)等。其中羟基脲(Hu)应用及实验研究较多。羟基脲(Hu)是一种低毒和可有效增加γ珠蛋白链和β珠蛋白链合成,从而导致血液学和临床症状明显改善,羟基脲(Hu)治疗的剂量及方法:5天疗法:50mg/(kg·d)×5天为1个疗程。10~30mg/(kg·d),连用3周为1个疗程,或25~50mg/(kg·d)×5~7天为1个疗程。有学者采用15~20mg/(kg·d)连续用药方法。主要对某些β地贫基因缺陷类型有效:28/654-2或-28/41-42双重杂合子,β-28纯合子。IVS-2-654 C→T突变中间型β-地贫。HbE/β-28双重杂合子。5~7天显效,Hb上升水平20~45g/L。
二、预后
β-地中海贫血重型并发症常是导致患儿死亡的重要原因,如不治疗,多于5岁前死亡。中间型和轻型地中海贫血正确处理可长期存活。
疾病护理:
小儿β地中海贫血一般护理
小儿β地中海贫血护理
1.地中海贫血患者往往体虚,要慎起居,适寒温,注意预防外感;多进行户外活动;呼吸新鲜空气;进行适宜的体育锻炼,气功锻炼,打太极拳等有助于增强体质和抗病能力。
2.饮食调理注意饮食调养。宜进食营养丰富的食物。
3.注意精神调养,对于虚劳血虚的预防有重要意义。七情过激,易使阴血暗耗,既是致病之因,又可加剧进程。所以本病患者应保持心情舒畅,乐观。
疾病饮食:
小儿β地中海贫血饮食原则
小儿β地中海贫血饮食
高蛋白低脂肪 :对一般贫血病人来说 ,首先应考虑给予高蛋白饮食.这可以通过食用动物的瘦肉以及肝,肾等内脏 ,获得优质蛋白的补充.其次 ,应尽量控制脂肪的摄入量.因为脂肪可抑制人体的造血功能 ,高脂肪还可导致腹泻,消化不良,肥胖病等疾患. 丰富的维生素 :饮食结构中维生素的含量丰富 ,对各类疾病的患者都是适宜的.就贫血病人而言 ,维生素 B 1,维生素 B 12,维生素 C和叶酸等是至关重要的.维生素 B 1的补充 ,可以通过粮食特别是粗杂粮食物获得 ;维生素 B 12和叶酸 ,主要来源于动物内脏等食物 ;维生素 C的主要来源 ,则是各种新鲜的蔬菜和水果.





