
肌张力障碍简介:肌张力障碍综合征(dystonic syndrome)简称肌张力障碍(dystonia),是主动肌与拮抗肌收缩不协调或过度收缩,引起以肌张力异常动作和姿势为特征的运动障碍综合征,具有不自主性和持续性特点。尽管名为肌张力障碍综合征,但肌张力的变化不引人注意,而引人注意的是异常的体位姿势和不自主的变换动作。肌张力障碍可影响受累肢体的正常运动幅度、范围、速度和肌肉的硬度等。
- 发病部位
- 传染性
- 治愈率
- 多发人群
- 相关症状
- 并发疾病
- 肌张力障碍综合征,肌紧张不足综合征,肌张力障碍综合症
- 肌肉
- 无传染性
- 70%
- 5~15岁,有家族史者
- 发作性睡眠,儿童型肌张力障碍,构音障碍
- 食管受压性吞咽困难,畸形性吞咽困难
- 挂号科室
- 治疗方法
- 治疗周期
- 治疗费用
- 临床检查
- 无
- 神经内科
- 病原治疗、药物治疗
- 10-30天
- 市三甲医院约(2000-10000元)
- 微量元素,血液电解质,MRI
- 常见药品
- 巴氯芬,左旋多巴,注射用A型肉毒毒素
- 在线购药
疾病症状:
肌张力障碍症状诊断
一、症状
肌张力障碍的临床分类目前尚不统一。肌张力障碍的各种分类与临床特点相关,主要根据肌张力障碍的受累肢体和部位,可能造成肌张力障碍的原因,发病时的年龄等。
1.国内王维治主编的《神经病学》(2006年第1版)介绍如下分类
(1)按病变累及部位分类:
①局限性肌张力障碍:影响躯体某部,如痉挛性斜颈、痉挛性发音困难(声带)、睑痉挛、口-下颌肌张力障碍、书写痉挛(一侧上肢)、职业性痉挛、足肌张力障碍等。
②节段性肌张力障碍:表现节段性分布特点,如颅-颈部肌张力障碍,上肢伴或不伴中轴、头颈部肌张力障碍,下肢伴或不伴躯干肌张力障碍,躯干-颈部(不影响头面部)肌张力障碍等。
③多部位肌张力障碍:累及身体2个以上不靠近部位。
④广泛性肌张力障碍:影响广泛的躯体范围。
⑤偏侧性肌张力障碍:仅累及单侧身体。
(2)按病因分类:
①遗传性进行性肌张力障碍:多在小儿发病,表现步行障碍及运动减少,躯干姿势异常,腰部明显向前弯曲,日内症状波动明显,晨轻,午后至傍晚加重,呈进行性发展,L-dopa疗效显著,本病与Parkinson病可能有类似之处。
②症状性肌张力障碍:如脑性瘫痪、累及基底核的脑卒中、Wilson病、Hallervorden-Spatz病、脑炎、脑肿瘤、精神病药物副反应等伴肌张力障碍,Parkinson病有时可伴肌张力障碍。
2.上海史玉泉等主编的《实用神经病学》(2004年第3版)则主张以下分类
(1)按肌张力障碍范围的分类:
①局限性肌张力障碍(focal dystonia):仅累及眼睑部肌群者,称眼睑痉挛(blepharospasm);累及口周及下颌肌群者,则称为口下颌肌张力障碍(oromandibular dystonia);疾病累及喉部肌群称为痉挛性构音障碍(spasmodic dysphonia);疾病累及颈部肌群称为痉挛性斜颈(spasmodic torticollis);疾病累及前臂和手部,称为书写性痉挛症(writer cramp)。
②节段性肌张力障碍(segmental dystonia):累及颅和颈部两处肌群者,称之为颅部节段性肌张力障碍(cranial segmental dystonia);累及颈和躯干者称为纵轴节段性肌张力障碍(axial segmental dystonia);累及一侧手臂和纵轴躯干或双上臂者,称为臂部节段性肌张力障碍(brachial segmental dystonia);累及一侧股肌和躯干或两侧股肌(可有或无躯干受累)者称为股节段性肌张力障碍(crural segmental dystonia)。
③偏身肌张力障碍(hemidystonia):系指同侧上下肢的肌群受累,多为继发性原因所造成的。
④全身肌张力障碍(generalized dystoina):系指疾病累及3个或3个以上肢体伴躯干、颅、颈或延髓部肌群,如全身性的扭转痉挛(torsion spasm)。
(2)按肌张力障碍起病年龄分类:
①儿童型肌张力障碍:患者年龄小于12岁。
②少年型肌张力障碍:患者年龄在13~20岁。
③成年型肌张力障碍:患者年龄大于20岁。
儿童期起病的肌张力障碍,病情常呈进行性,首先累及下肢,出现步态障碍,以后导致全身性肌张力障碍,预后较差。成年型肌张力障碍常为局限性,如颈、上肢,很少累及全身,腿部不受累,病情不进行性加重。
(3)按肌张力障碍病因分类:
①原发性肌张力障碍:
A.遗传性(常染色体显性或隐性遗传、X性连锁隐性遗传):肌阵挛性肌张力障碍(酒精反应性、常染色体显性遗传),发作性肌张力障碍(paroxysmal dystonia),发作性睡眠性肌张力障碍(paroxysmal hypnogenetic dystonia),典型“特发性”扭转型肌张力障碍(单纯肌张力障碍、常染色体显性遗传、dytl基因),非典型“特发性”扭转型肌张力障碍(常染色体显性遗传),不典型肌张力障碍(常染色体显性遗传)。
B.散发性肌张力障碍。
②继发性肌张力障碍:包括多巴反应性肌张力障碍(基因位于染色体14),散发性肌张力障碍伴帕金森综合征,X-性连锁隐性遗传肌张力障碍-帕金森综合征(菲律宾人中),遗传性肌阵挛性肌张力障碍,少年型亨廷顿舞蹈病,家族性肌萎缩性肌张力障碍性截瘫,哈勒沃登-施帕茨(Hallervorden-Spatz)病,进行性苍白球变性,遗传性共济失调,肌张力障碍伴神经性聋,约瑟夫(Joseph)病,共济失调毛细血管扩张症,Rett综合征,婴儿双侧纹状体坏死,家族性基底核钙化,神经棘红细胞增多症等。也有酚噻嗪类药、颅脑外伤、基底核肿瘤、代谢性疾病造成。
3.肌张力障碍的主要临床特点
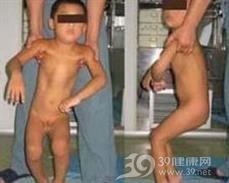
多于10~20岁发病,初期进展较快,尔后逐渐缓慢,有时不进展或稍改善,病程不定。日本散发病例较多,其他国家家族性发病较多。临床表现症状体征多样,如扭转痉挛样运动、痉挛性斜颈、书写痉挛、投掷运动和姿势异常,步行姿势不自然等。初期立位或动作时常不自主出现姿势或肢体位置异常,卧位时肌紧张消失。随病程进展,卧位也出现异常肌紧张,可见骨变形或肌肉异常发育,粗大动作时各肌肉肌力虽无明显异常,但因肌紧张不能完成精巧动作,但仍可写字、步行和骑自行车,有时可伴手指震颤或肌阵挛动作等。
二、诊断
特发性痉挛性斜颈是颈肌肌张力障碍的表现。书写痉挛(writer cramp)是执笔书写时手和前臂出现肌张力障碍姿势,表现握笔如握匕首、手臂僵硬、手腕屈曲、肘部不自主地向外弓形抬起、手掌面向侧面等,但做其他动作正常。也可表现其他职业性痉挛,如弹钢琴、打字,使用螺丝刀或餐刀等。药物治疗通常无效,可让病人学会用另一只手完成这些任务。
对扭转痉挛、痉挛性斜颈、书写痉挛、投掷运动等肌张力障碍患者较易诊断。确定本综合征后,还应查找病因。
疾病病因:
肌张力障碍疾病病因
一、发病原因
特发性肌张力障碍(idiopathic dystonia)病因不明,可能与遗传有关,继发性肌张力障碍常是基底核、丘脑及脑干网状结构等病变的症候。
二、发病机制
1、特发性肌张力障碍(idiopathic dystonia)可为常染色体显性(30%~40%外显率)、常染色体隐性或X连锁隐性遗传,显性遗传缺损基因DYT1已定位于9号常染色体长臂9q32~34,编码ATP结合蛋白扭转蛋白A(torsin A),可有散发病例。环境因素如创伤或过劳等可诱发,如口-下颌肌张力障碍发病前,可有面部或牙损伤史,一侧肢体过劳,常诱发书写痉挛、打字员痉挛和运动员肢体痉挛等。
2、继发性肌张力障碍是纹状体、丘脑、蓝斑、脑干网状结构等病变所致,如肝豆状核变性、胆红素脑病、神经节苷脂沉积症、苍白球黑质红核色素变性、进行性核上性麻痹、特发性基底核钙化、甲状旁腺功能低下、中毒、脑卒中、脑外伤、脑炎等;另外,药物(左旋多巴、吩噻嗪类、丁酰苯类、甲氧氯普胺)也可诱发。
肌张力障碍还可由心理因素引起,特点是易受到暗示影响。
3、病理及神经生化改变:特发性扭转痉挛可见非特异性病理改变,包括壳核、丘脑及尾状核小神经元变性,基底核脂质及脂色素增多。继发性扭转痉挛病理学特征随原发病不同而异,痉挛性斜颈、Meige综合征、书写痉挛和职业性痉挛等局限性肌张力障碍无特异性病理改变。
4、拮抗肌过度协同性收缩是本病主要的生理学特点,肢体每次收缩常由近端向远程扩展。肌电图检查发现,肌肉收缩间歇期无不自主运动电位。根据肌电图特点分为3型:
①持续30s的肌肉收缩,间隔短时间静息;
②重复、节律性收缩和静息状态,收缩期和静息期均为1~2秒钟;
③快速、短暂肌肉收缩,持续100ms,表现类似肌阵挛。
疾病预防:
肌张力障碍预防
有遗传背景的疾病,预防显得更为重要。预防措施包括避免近亲结婚,推行遗传咨询、携带者基因检测及产前诊断和选择性人工流产等,防止患儿出生。早期诊断、早期治疗、加强临床护理,对改善患者的生活质量有重要意义。
疾病鉴别:
肌张力障碍鉴别诊断
一、鉴别
肌张力障碍综合征须注意与肌阵挛、癔症等鉴别。本病的肌紧张是中枢性较剧烈的不随意肌收缩,注意与Parkinson病肌强直不同。
疾病检查:
1.核磁共振成像(MRI)
核磁共振成像也称磁共振成像,是利用核磁共振原理,通过外加梯度磁场检测所发射出的电磁波,据此可以绘制成物体内部的结构图像。
2.CT检查
CT是一种功能齐全的病情探测仪器,它是电子计算机X射线断层扫描技术简称。CT检查是根据人体不同组织对X线的吸收与透过率的不同,应用灵敏度极高的仪器对人体进行测量,然后将测量所获取的数据输入电子计算机,电子计算机对数据进行处理后,就可摄下人体被检查部位的断面或立体的图像,发现体内任何部位的细小病变。
3.正电子发射计算机断层扫描(PET)
正电子发射型计算机断层显像(Positron Emission Computed Tomography,简称PET),是核医学领域比较先进的临床检查影像技术。 正常范围PET特别适用于在没有形态学改变之前,早期诊断疾病,发现亚临床病变以及评价治疗效果。目前,PET在肿瘤、冠心病和脑部疾病这三大类疾病的诊疗中尤其显示出重要的价值。
4.基因分析
对确诊某些遗传性肌张力障碍疾病有重要意义。
疾病就诊:
1、描述就诊原因(从什么时候开始,有什么不舒服?)
2、不适的感觉是否由明显的因素引起?
3、有痉挛性斜颈、书写痉挛等伴随症状?
4、是否到过医院就诊,做过那些检查,检查结果是什么?
5、治疗情况如何?
6、有无药物过敏史?
疾病治疗:
肌张力障碍一般治疗
肌张力障碍西医治疗
一、治疗
1.病因治疗 如Wilson病驱铜治疗及调整饮食可控制症状,药物诱导肌张力障碍可停药或换药治疗。
2.对症治疗 药物诱导急性肌张力障碍用苯海拉明(diphenhydramine)50mg或地西泮(安定)5~10mg缓慢静脉注射,可快速缓解症状;也可口服地西泮(安定)10~20mg/d,苯海索(三己芬迪)6~30mg/d,L-dopa 1~3g/d,氟哌啶醇(holoperidol)1.5~4.5mg/d,可使约50%的病例症状缓解。生活指导及生物反馈疗法也有一定的治疗价值。改善症状药物种类虽较多,如地西泮(安定)、劳拉西泮(罗拉)、苯海索(安坦)、扑米酮(扑痫酮)、丙戊酸钠(丙戊酸)、卡马西平、巴氯芬(脊舒)、多巴胺受体激动药、左旋多巴和锂剂等,但无任何一种药物对所有慢性肌张力障碍都有效,临床应对不同患者通过反复疗效观察,寻找有效药物和最佳治疗剂量。
3.肉毒毒素A 局限性肌张力障碍受累肌肉在肌电图引导下注射肉毒毒素A有效率达95%,手部及颈部有效率85%,可维持3~4个月,反复注射疗效维持时间缩短。
4.外科治疗 药物及肉毒毒素A注射无效时可选择手术,神经切断术可解除血管对神经压迫,广泛性及偏侧性肌张力障碍可选用丘脑损毁术。
二、预后
不同类型预后不尽相同,一般为良性过程,如原发性书写痉挛症状相当稳定,很少有扩散加重倾向。
疾病护理:
肌张力障碍一般护理
肌张力障碍护理
调整日常生活与工作量,有规律地进行活动和锻炼,避免劳累。
2.保持情绪稳定,避免情绪激动和紧张。
3.避免寒冷刺激,注意保暖。
疾病饮食:
肌张力障碍饮食原则
肌张力障碍饮食保健
宜吃芹菜、金针菜、韭菜、冬瓜、乌梅、柿饼、芝麻、莲子、海参。





