
先天性厚甲症简介:先天性厚甲症(pachyonychia congenita)是一种少见的甲肥厚性遗传性皮肤病,于1906年由Jadassohn和Lewandowsky首先命名。分类:通常分为四型,Ⅰ型最常见,为Jadassohn-Lewandowsky综合征;Ⅱ型为Jackson-Sertoli综合征;Ⅲ型又名为Schaferer-Branauer综合征;Ⅳ型又名迟发型先天性厚甲症(pachyonychia congenita tarda)。
- 发病部位
- 传染性
- 治愈率
- 多发人群
- 相关症状
- 并发疾病
- 皮肤
- 无传染性
- 50%,以对症治疗为主
- 所有人群
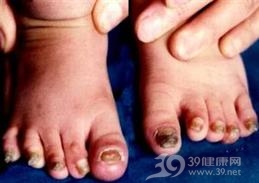
- 指甲嵌入肉,掌跖角化过度,囊肿
- 黏膜白斑
- 挂号科室
- 治疗方法
- 治疗周期
- 治疗费用
- 临床检查
- 有
- 手外科,儿科
- 药物治疗
- 1-3年
- 市三甲医院约(1000-3000元)
- 免疫病理检查,皮肤病的物理检查,皮肤涂片显微镜检查
- 常见药品
- 小儿腹泻散
- 在线购药
疾病症状:
一、症状
Ⅰ型先天生厚甲症的特征性表现为指(趾)甲显著肥厚,掌跖角化过度、多汗,肘膝臀部出现毛囊角化过度性皮损等。其中甲改变于出生后出现,但更多见于出生后几个月内,因角化过度导致甲远端向上翘起,出现特征性的门槛样远端角化过度。黏膜白斑多见于舌和口腔,偶尔累及喉部。引起声嘶。此外于足跖受压部可见胼胝,行走可导致水疱。手掌胼胝仅见于手工劳动者。
其他各型临床表现与Ⅰ型类似,但Ⅱ型伴发胎生牙,多发性皮脂腺囊肿;Ⅲ型伴发角膜白斑;Ⅳ型发病较晚,于颈、腰、腋窝、大腿、膝的屈侧、臀部和腹部可见色素沉着。
二、诊断
根据临床表现,皮损特点,组织病理特征性即可诊断。
疾病病因:
一、发病原因
为常染色体显性遗传病。发病机制还不很清楚。
二、发病机制
为常染色体显性遗传病。Ⅰ型与角蛋白16和6a的突变有关,Ⅱ型与角蛋白17和6b的突变有关。
疾病预防:
本病暂无有效预防措施,早发现早诊断是本病防治的关键。
疾病鉴别:
目前没有相关内容描述。
疾病检查:
组织病理显示色素失禁和淀粉样物质沉积。
疾病就诊:
疾病治疗:
先天性厚甲症一般治疗
一、治疗
对厚甲行拔除术只能暂时缓解症状。去除甲母质无益于疗效。但甲母质和甲床刮除术为简单且有效的疗法。角化性皮损可局部应用角质溶解剂,如乳酸洗液,水杨酸和尿素制剂等。维A酸(异维甲酸)口服能清除角化性丘疹和黏膜白斑,但对掌跖角化无效,迟发型先天性厚甲症患者可试用activetin治疗。
二、预后
Ⅳ型发病较晚,于颈、腰、腋窝、大腿、膝的屈侧、臀部和腹部可见色素沉着。
疾病护理:
疾病饮食:
先天性厚甲症饮食原则
1、饮食上应注意清淡,多以菜粥、面条汤等容易消化吸收的食物为佳。
2、可多食新鲜的水果和蔬菜,以保证维生素的摄入量。
3、给予流质或半流质的食物,如各种粥类、米汤等。





