
帕金森简介:帕金森病(Parkinson's disease)又称"震颤麻痹",巴金森氏症或柏金逊症,多在60岁以后发病。主要表现为患者动作缓慢,手脚或身体其它部分的震颤,身体失去柔软性,变得僵硬。最早系统描述该病的是英国的内科医生詹母帕金森,当时还不知道该病应归入哪一类疾病,称该病为震颤麻痹。帕金森病是老年人中第四位最常见的神经变性疾病,在65岁人群中,1%患有此病;在>40岁人群中则为0.4%.本病也可在儿童期或青春期发病。50%~80%的病例起病隐袭,首发症状通常是一侧手部的4~8Hz的静止性"捻丸样"震颤。言语障碍是帕金森病患者常见症状。
- 发病部位
- 传染性
- 治愈率
- 多发人群
- 相关症状
- 并发疾病
- 帕金森病,震颤麻痹,特发性帕金森病
- 颅脑 全身
- 无传染性
- 老年人,于40~70岁,60岁后发...
- 静止性震颤,肌张力过高,感觉障碍
- 褥疮,感冒,便秘
- 挂号科室
- 治疗方法
- 治疗周期
- 治疗费用
- 临床检查
- 无
- 神经内科,中医科
- 药物治疗
- 5000-10000元)
- SPECT显像,非必需微量元素,大生化检查
- 常见药品
- 单唾液酸四己糖神经节苷脂钠注射液,注射用单唾液酸四己糖神经节苷脂钠,多巴丝肼片
- 在线购药
疾病症状:
帕金森症状诊断
一、症状:
帕金森病通常发病于40~70岁,60岁后发病率增高,30多岁前发病少见,在一组380例PD患者中仅4例;男性略多。起病隐袭,发展缓慢,主要表现静止性震颤、肌张力增高和运动迟缓等,症状出现孰先孰后因人而异。首发症状震颤最多(60%~70%),其次为步行障碍(12%)、肌强直(10%)和运动迟缓(10%)。症状常自一侧上肢开始,逐渐波及同侧下肢、对侧上肢及下肢,呈N字型进展(65%~70%);25%~30%的病例可自一侧下肢开始,两侧下肢同时开始极少见,不少病例疾病晚期症状仍存在左右差异。
但不论如何治疗,慢性进展性病程、数年后多数患者需要帮助是其固有的临床特点。根据PD的典型表现及对多巴药物的正性反应,一般可以做出明确的诊断。但是,对于某些亚临床症状或非典型病例在早期确难以认识,而早期确诊、早期治疗对后期生活质量有着重要影响,这也是目前临床学界研究的重点。对于大部分患者和临床医师来说,很难肯定和判定PD的发病日期、首发症状,以及确定动作缓慢、震颤症状出现的时间。据国内李大年等的报告,推测PD的临床前期症状可能有3~5年之久,为此,可将PD症状分为临床前期症状和临床期症状两个阶段。
1.临床前期症状
最早提出临床前期症状仅见于Fletcher(1973)等人的报告,但他们提出的这些症状至今尚未得到人们的重视。这些症状主要包括以下两方面:
(1)感觉异常:
事实上早在Parkinson《震颤麻痹》一书中就描述部分PD病例在其运动症状出现之前可出现风湿样疼痛,同年Charcot也对2例PD患者作了同样的描述。直到20世纪70年代,Fletcher和Snider等人才对PD的临床前期症状及感觉障碍作了比较详细的描述。到20世纪80年代,William等人结合电生理学对感觉障碍进行了分类,他报告的感觉症状主要表现为患肢关节处无缘由的麻木、刺痛、蚁行感和烧灼感,以腕、踝处为主,开始多为间歇性或游走性,后期表现为固定性。常规神经系统查体无明显客观感觉异常,电生理检查可见部分病例的体感诱发电位(SEP),特别是下肢的潜伏期和传导时间延长。到20世纪90年代初,我们对150例患者作了回顾性调查,结果是全部患者不同程度的在PD临床症状出现前体验过患肢感觉异常,而且这种异常可一直持续下去,但与运动障碍不成平行关系。电生理检查主要是体感、皮质诱发电位有皮质延搁(centre delay)和传导延迟及潜伏期延长。
(2)不宁肢与易疲惫:
除主观感觉异常外,约1/2患者在早期曾体验过患肢难以描述的酸、胀、麻木或疼痛等不适感,而且这种不适感多在劳累后的休息时发生或明显,经敲、捶打后可缓解,酷像不宁腿综合征的表现。另则,部分患者的患肢易出现疲劳感,特别是上肢的腕关节、肩关节,下肢的踝关节和膝关节,当劳累后这些部位可出现难以发现的轻微震颤。对这些症状开始时服用一般镇痛药可有效,数月后则无作用。此时服用多巴药物后可出现明显疗效。
2.临床期症状
首发症状存在着明显个体差异,有报告统计主观感觉异常为85%、震颤为70.5%、肌僵直或动作缓慢为19.7%、失灵巧和(或)写字障碍为12.6%、步态障碍为11.5%、肌痛痉挛和疼痛为8.2%、精神障碍如抑郁和焦虑紧张等为4.4%、语言障碍为3.8%、全身乏力或肌无力为2.7%、流口水和面具脸各为1.6%。
(1)静止性震颤(static tremor):
常为PD首发症状,少数患者尤其70岁以上发病者可不出现震颤。其机制是受累肌群与拮抗肌群规律性、交替性不协调活动所致。早期常表现在肢体远端,始于一侧,以上肢的手部震颤为多见,部分患者始于下肢的膝部。当伴有旋转的成分参与时,可出现拇指、示指搓丸样震颤。震颤频率一般在4~8Hz,静止时出现,大力动作时停止,紧张时加剧,睡眠时消失。经数年后累及到同侧上下肢或对侧,严重者可出现头部、下颌、口唇、舌、咽喉部以及四肢震颤。令患者活动一侧肢体如握拳或松拳,可引起另侧肢体出现震颤,该试验有助于发现早期轻微震颤。后期除静止性震颤外,部分患者可合并动作性或姿势性震颤。
(2)肌强直(rigidity):
肌强直是PD的主要症状之一,主要是由于主动肌和拮抗肌均衡性张力增高所致。如果在被动运动中始终存在,则被称之为铅管样强直或张力,若同时伴有震颤时,被动运动时可感到有齿轮样感觉,则称之为齿轮样强直或张力。肌强直最早发生在患侧的腕、踝,特别是患者劳累后,轻缓的被动运动腕、踝关节时可感到齿轮样肌张力增高。由于肌张力的增高,可给患者带来一系列的异常症状,如瞬目、咀嚼、吞咽、行走等动作减少。
以下临床试验有助于发现轻微肌强直:
①令患者运动对侧肢体,被检肢体肌强直可更明显;
②头坠落试验(head dropping test):患者仰卧位,快速撤离头下枕头时头常缓慢落下,而非迅速落下;
③令患者把双肘置于桌上,使前臂与桌面成垂直位,两臂及腕部肌肉尽量放松,正常人此时腕关节与前臂约成90度屈曲,PD患者腕关节或多或少保持伸直,俨若竖立的路标,称为“路标现象。老年患者肌强直引起关节疼痛,是肌张力增高使关节血供受阻所致。
(3)运动迟缓(bradykinesia):
表现随意动作减少,包括始动困难和运动迟缓,因肌张力增高、姿势反射障碍出现一系列特征性运动障碍症状,如起床、翻身、步行和变换方向时运动迟缓,面部表情肌活动减少,常双眼凝视,瞬目减少,呈面具脸(maskedface),手指精细动作如扣纽扣、系鞋带等困难,书写时字愈写愈小,为写字过小征(micrographia)等。
PD患者的运动缓慢或不能是致残的主要原因。过去认为PD的运动不能是由于肌强直所致,事实上两者并无因果关系。现已初步证明,PD的运动减少和不能是一个很复杂的症状,它主要和皮质下锥体外系的驱动装置功能或锥体外系下行运动激活装置障碍有关。因为对运动不能的患者进行手术治疗后,肌强直症状明显改善,但其运动频度并非像服用多巴药物后成一致性改善。
(4)姿势步态异常:
姿势反射障碍是带给PD患者生活困难的主要症状,它仅次于运动减少或运动不能。患者四肢、躯干和颈部肌强直呈特殊屈曲体姿,头部前倾,躯干俯屈,上肢肘关节屈曲,腕关节伸直,前臂内收,指间关节伸直,拇指对掌;下肢髋关节与膝关节均略呈弯曲,早期下肢拖曳,逐渐变为小步态,起步困难,起步后前冲,愈走愈快,不能及时停步或转弯,称之为慌张步态(festination),行走时上肢摆动减少或消失;转弯时因躯干僵硬,躯干与头部联带小步转弯,与姿势平衡障碍导致重心不稳有关。患者害怕跌倒,遇小障碍物也要停步不前。随疾病进展姿势障碍加重,晚期自坐位、卧位起立困难。目前对PD患者这种固有的姿势反射障碍的机制尚无明确解释,有人认为该症状主要与苍白球经丘脑至皮质的传出环路损害有关。
(5)其他症状:
①反复轻敲患者眉弓上缘可诱发眨眼不止(Myerson征),正常人反应不持续;可有眼睑阵挛(闭合眼睑轻度颤动)或眼睑痉挛(眼睑不自主闭合)。
②口、咽、腭肌运动障碍,使讲话缓慢,语音低沉单调,流涎等,严重时四、PD的分类:
(1)原发性(特发性帕金森病,即震颤麻痹):
①按病程分型:
A.良性型:病程较长,平均可达12年运动症状波动和精神症状出现较迟。
B.恶性型:病程较短,平均可达4年。运动症状波动和精神症状出现较早。
②按症状分型:
A.震颤型。
B.少动和强直型。
C.震颤少动和强直型伴痴呆型。
D.震颤少动和强直型不伴痴呆型。
③按遗传分型:
A.家族性帕金森病。
B.少年型帕金森病。
(2)继发性(帕金森综合征、症状性帕金森综合征):
①感染性(包括慢性病毒感染)脑炎后帕金森综合征(嗜睡性脑炎、其他脑炎等)。
②中毒性(一氧化碳、锰、二硫化碳、氢化物、甲醇等)。
③药物性(抗精神病药物如吩噻嗪类、丁酰苯类、萝芙木生物碱及α-甲基多巴等)。
④脑血管病变。
⑤脑肿瘤(特别是脑部中线肿瘤)。
⑥脑外伤。
⑦中脑空洞。
⑧代谢性(甲状腺功能减退、基底节钙化、慢性肝脑变性等)。
(3)症状性帕金森综合征(异质性系统变性):
①进行性核上性麻痹。
②纹状体黑质变性。
③皮质齿状核黑质变性。
④橄榄脑桥小脑萎缩。
⑤Shy-Drager位置性低血压综合征。
⑥痴呆[关岛帕金森-痴呆-肌萎缩性侧索硬化综合征,Jacob-Creutfeldt病(皮质纹状体脊髓变性),Alzheimer及Pick病,正常颅压脑积水]。
⑦遗传性疾病(肝豆状核变性、Hallerrorden-Spatz病、Huntington病、脊髓小脑黑质变性等)。
二、诊断
1.诊断依据
(1)中老年发病,缓慢进行性病程。
(2)四项主征(静止性震颤、肌强直、运动迟缓、姿势步态障碍)中至少具备2项,前两项至少具备其中之一,症状不对称。
(3)左旋多巴治疗有效,左旋多巴试验或阿朴吗啡试验阳性支持原发性PD诊断。
(4)患者无眼外肌麻痹、小脑体征、体位性低血压、锥体系损害和肌萎缩等。PD临床诊断与尸检病理证实符合率为75%~80%。
2.国内外常用的诊断与鉴别诊断标准
(1)原发性帕金森病(IPD)的诊断:
王新德执笔1984年10月全国锥体外系会议制定的标准如下:
①至少要具备下列4个典型的症状和体征(静止性震颤、少动、僵直、位置反射障碍)中的2个。
②是否存在不支持诊断IPD的不典型症状和体征,如锥体束征、失用性步态障碍、小脑症状、意向性震颤、凝视麻痹、严重的自主神经功能障碍、明显的痴呆伴有轻度锥体外系症状。
③脑脊液中高香草酸减少,对确诊早期帕金森病(PD)和特发性震颤(ET)、药物性帕金森综合征与PD是有帮助的。
一般而言,ET有时与早期IPD很难鉴别,ET多表现为手和头部位置性和动作性震颤而无肌张力增高和少动。
(2)继发性帕金森综合征(SPDS)的诊断:
①药物性PS(MPS):药物性PS与IPD在临床上很难区别,重要的是依靠是否病史上有无服用抗精神病药物史。另外,药物性PS的症状两侧对称,有时可伴有多动症侧会先出现症状。若临床鉴别困难时,可暂停应用抗精神病药物,假若是药物性,一般在数周至6个月PS症状即可消失。
②血管性PS(VPS):该征的特点为多无震颤,常伴有局灶性神经系统体征(如锥体束征、假性球麻痹、情绪不稳等),病程多呈阶梯样进展,L-多巴制剂治疗一般无效。
(3)症状性帕金森病综合征(异质性系统变性)的诊断:
①进行性核上性变性:有时与帕金森病很难鉴别。进行性核上性麻痹的临床特点主要为动作减少,颈部强直并稍后仰及假性延髓麻痹和向上凝视麻痹。
②橄榄脑桥小脑萎缩:原发性帕金森病应与本病进行鉴别。橄榄脑桥小脑变性临床也可表现为少动、强直、甚至静止性震颤。但多同时有共济失调等小脑症状。CT检查亦可见特征性的改变。血谷氨酸脱羧酶活力减低。
③纹状体黑质变性:本病与原发性帕金森病很想象,临床上很难鉴别,主要依靠病理诊断。若临床上L-多巴治疗无效时,应考虑纹状体黑质变性可能。
④Shy-Drager位置性低血压综合征:临床表现为位置性低血压、大小便失禁、无汗、肢体远端小肌肉萎缩等。有时也可伴有帕金森病综合征。若临床发现患者有帕金森病综合征和轻度自主神经障碍症状,就需要与原发性帕金森病鉴别。
⑤痴呆:痴呆伴有帕金森综合征不罕见。A.Alzheimer病:晚期Alzheimer病除痴呆外,尚有锥体外系症状,如少动、强直和口面多动。另外由于帕金森病甚至早期也可伴有痴呆,因此需依靠随访对两者进行鉴别;B.正常颅压脑积水:本病表现为步态障碍、尿失禁和痴呆。有时也可出现帕金森病的症状,如少动、强直、和静止性震颤等。CT检查对鉴别有帮助。放射性核素脑池造影对诊断正常颅压脑积水也有重要意义。
⑥遗传变性疾病:
A.苍白球-黑质色素变性病(Hallervorden-Spatz disease)。
B.Huntinton舞蹈病。
C.Lubag(X-连肌张力失常-PDS)。
D.线粒体细胞病伴纹状体坏死。
E.神经棘红细胞增多症(beta-脂蛋白缺乏症)。
F.肝豆状核变性(Wilson病)。
原发性PD在这些临床类型中占总数75%~80%;继发性(或症状性)PD相对少见;遗传变性病与帕金森叠加综合征占10%~15%。
对大多数已有明显的动作缓慢、减少、肌强直、震颤的中老年患者均会被考虑到IPD,而对那些早期或症状不典型的病例有时确会被误诊。为此,Takahashi等(1992)和Calne等(1992)提出原发性帕金森病(IPD)早期诊断的必要条件和删除条件的初步标准(表1、2)。
疾病病因:
帕金森疾病病因
(一)发病原因
特发性帕金森病(idiopathic Parkinson’s disease)病因迄今未明。某些中枢神经系统变性疾病伴Parkinson病症状,以中枢神经系统不同部位变性为主,尚有其他临床特点,故可称之为症状性Parkinson病,如进行性核上性麻痹(PSP)、纹状体黑质变性(SND)、Shy-Drager综合征(SDS)及橄榄脑桥小脑萎缩(OPCA)等。还有一些疾病或因素可以产生类似PD临床症状,其病因为感染、药物(多巴胺受体阻滞药等)、毒物(MPTP、一氧化碳、锰等)、血管性(多发性脑梗死)及脑外伤等所致,临床上称为帕金森综合征(Parkinson’s syndrome,Palkinsonism)。
迄今为止,PD的病因仍不清楚。目前的研究倾向于与年龄老化、遗传易感性和环境毒素的接触等综合因素有关。
1)年龄老化:帕金森主要发生于中老年人,40岁以前发病少见,提示老龄与发病有关。研究发现,自30岁以后,黑质多巴胺能神经元、酪氨酸氧化酶和多巴脱羧酶活力,纹状体多巴胺递质水平随年龄增长逐渐减少。然而,仅少数老年人患此病,说明生理性多巴胺能神经元蜕变不足以致病,年龄老化只是本病发病的促发因素。
2)环境因素:流行病学调查结果发现,帕金森病的患病率存在地区差异,所以人们怀疑环境中可能存在一些有毒的物质,损伤了大脑的神经元。
3)遗传易患性。近年在家族性帕金森病患者中曾发现a共同核素基因的Alα-53THr突变。但以后多次未被证实。
4)家族遗传性:医学家们在长期的实践中发现帕金森病似乎有家族聚集的倾向,有帕金森病患者的家族其亲属的发病率较正常人群高一些。
目前普遍认为,帕金森并非单一因素,多种因素可能参于其中。遗传因素可使患病易感性增加,只有与环境因素及衰老的相互作用下,通过氧化应激、线粒体功能衰竭、钙超载、兴奋性氨基酸毒性作用、细胞凋亡、免疫异常等机制才导致黑质多巴胺能神经元大量变性丢失而发病。
(二)发病机制
1.发病机制 十分复杂,可能与下列因素有关。
(1)年龄老化:
PD主要发生于中老年,40岁前发病少见,提示老龄与发病有关。研究发现自30岁后黑质DA能神经元、酪氨酸羟化酶(TH)和多巴脱羧酶(DDC)活力、纹状体DA递质逐年减少,DAD1和D2受体密度减低。但老年人患PD毕竟是少数,说明生理性DA能神经元退变不足以引起本病。实际上,只有黑质DA能神经元减少50%以上,纹状体DA递质减少80%以上,临床才会出现PD症状,老龄只是PD的促发因素。
(2)环境因素:
流行病学调查显示,长期接触杀虫剂、除草剂或某些工业化学品等可能是PD发病危险因素。20世纪80年代初美国加州一些吸毒者因误用一种神经毒物质吡啶类衍生物1-甲基4-苯基1,2,3,6-四氢吡啶(MPTP),出现酷似原发性PD的某些病理变化、生化改变、症状和药物治疗反应等,给猴注射MPTP也出现相似效应。嗜神经毒MPTP和某些杀虫剂、除草剂可能抑制黑质线粒体呼吸链NADH-CoQ还原酶(复合物Ⅰ)活性,使ATP生成减少,自由基生成增加,导致DA能神经元变性死亡。PD黑质区存在明显脂质过氧化,还原型谷胱甘肽显著降低,提示抗氧化机制障碍及氧化应激可能与PD有关。
(3)遗传因素:
约10%的患者有家族史,呈不完全外显的常染色体显性遗传或隐性遗传,其余为散发性PD。双胞胎一致性研究显示,某些年轻(<40岁)患者遗传因素可能起重要作用。迄今已确定PARK 1~10等10个单基因与PD有关,其中已确认三个基因产物与家族性PD有关:
①alpha;-突触核蛋白为PARK1基因突变,基因定位于4号染色体长臂4q21~23,alpha;-突触核蛋白可能会增高DA能神经细胞对神经毒素敏感性;
②Parkin为PARK2基因突变,定位于6号染色体长臂6q25.2~27;
③泛素蛋白C末端羟化酶-L1为PARK5基因突变,定位于4号染色体短臂4p14。细胞色素P45O2D6基因和某些线粒体DNA突变可能是PD发病易感因素之一,可能使P450酶活性下降,使肝脏解毒功能受损,易造成MPTP等毒素对黑质纹状体损害。
(4)氧化应激和自由基生成:
自由基可使不饱和脂肪酸发生脂质过氧化(LPO),后者可氧化损伤蛋白质和DNA,导致细胞变性死亡。PD患者由于B型单胺氧化酶(MAO-B)活性增高,可产生过量OH基,破坏细胞膜。在氧化同时,黑质细胞内DA氧化产物聚合形成神经黑色素,与铁结合产生Fenton反应可形成OH。正常情况下,细胞内有足够的抗氧化物质,如脑内的谷胱甘肽(GSH)、谷胱甘肽过氧化物酶(GSH-PX)和超氧化物歧化酶(SOD)等,DA氧化产生自由基不会产生氧化应激,保证免遭自由基损伤。PD患者黑质部还原型GSH降低和LPO增加,铁离子(Fe2 )浓度增高和铁蛋白含量降低,使黑质成为易受氧化应激侵袭的部位。
(5)线粒体功能缺陷:
近年发现,线粒体功能缺陷在PD发病中起重要作用。对PD患者线粒体功能缺陷认识源于对MPTP作用机制研究,MPTP通过抑制黑质线粒体呼吸链复合物Ⅰ活性导致Parkinson病。体外实验证实MPTP活性成分MPP 能造成MES 23.5细胞线粒体膜电势下降,氧自由基生成增加。PD患者黑质线粒体复合物Ⅰ活性可降低32%~38%,复合物α活性降低使黑质细胞对自由基损伤敏感性显著增加。在多系统萎缩及进行性核上性麻痹患者黑质中未发现复合物Ⅰ活性改变,表明PD黑质复合物Ⅰ活性降低可能是PD相对特异性改变。PD患者存在线粒体功能缺陷可能与遗传和环境因素有关,研究提示PD患者存在线粒体DNA突变,复合物Ⅰ是由细胞核和线粒体两个基因组编码翻译,两组基因任何片段缺损都可影响复合物Ⅰ功能。
(6)兴奋性毒性作用:
有作者应用微透析及HPLC检测发现,由MPTP制备的PD猴模型纹状体中兴奋性氨基酸(谷氨酸、天门冬氨酸)含量明显增高。若细胞外间隙谷氨酸浓度异常增高,会过度刺激受体,对CNS产生明显毒性作用。动物实验发现,脑内注射微量谷氨酸可导致大片神经元坏死,谷氨酸神经毒作用是通过受体起作用,NMDA受体介导兴奋性神经毒作用与DA能神经元变性有关。谷氨酸可通过激活NMDA受体产生一氧化氮(NO)损伤神经细胞,并释放更多兴奋性氨基酸,进一步加重神经元损伤。
(7)钙的细胞毒作用:
人类衰老可伴神经细胞内游离Ca2 浓度增加、Ca2 /Mg2 -ATP酶活性降低,线粒体储钙能力降低等。细胞内Ca2 浓度变化影响神经元多项重要功能,如细胞骨架维持、神经递质功能、蛋白质合成及Ca2 介导酶活性等,钙结合蛋白尤其28KD维生素D依赖性钙结合蛋白(Calbindin-D28K)可能扮演重要角色,与钙/镁-ATP酶激活有关,具有神经保护作用。Icopini和Christakos等报道,PD患者黑质、海马、缝背侧核Calbindin-D28K含量及mRNA表达明显低于正常人,提示钙结合蛋白基因表达降低也可导致细胞毒作用。
(8)免疫学异常:
Abramsky(1978)提出PD发病与免疫异常有关。临床研究发现PD患者细胞免疫功能降低,白细胞介素-1(IL-1)活性降低明显。McRae-Degueurce等报道PD患者脑脊液(CSF)存在抗DA能神经元抗体。细胞培养发现,PD血浆及CSF抑制大鼠中脑DA能神经元功能及生长。将PD患者血IgG立体定向注入大鼠一侧黑质,黑质酪氨酸羟化酶(TH)及DA能神经元明显减少,提示可能启动或参与免疫介导的黑质细胞损伤。肿瘤坏死因子α(TNF-α)、IL-6、上皮生长因子(EGF)、转移生长因子-α(TGF-α)和β2-微球蛋白(β2-MG)等可能与PD发病有关。
(9)细胞凋亡:
研究表明,PD发病过程存在细胞凋亡,自由基、神经毒素及神经营养因子缺乏等。Agid(1995)检测PD患者黑质DA能神经元凋亡形态学和生化特征,发现PD患者脑内约5能神经元有细胞凋亡特征性病变,存在TNF-α受体(α-TN-FR)和bcl-2原癌基因表达,细胞凋亡可能是DA能神经元变性的基本步骤。
目前普遍认为,PD并非单一因素致病,可能多种因素参与。遗传因素使患病易感性增加,在环境因素及年龄老化共同作用下,通过氧化应激、线粒体功能衰竭、钙超载、兴奋性氨基酸毒性及细胞凋亡等机制引起黑质DA能神经元变性,导致发病。
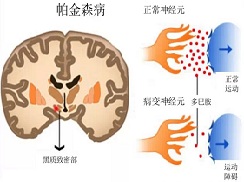
2.病理改变
PD主要病变是含色素神经元变性、缺失,黑质致密部DA能神经元最显著。镜下可见神经细胞减少,黑质细胞黑色素消失,黑色素颗粒游离散布于组织和巨噬细胞内,伴不同程度神经胶质增生。正常人黑质细胞随年龄增长而减少,黑质细胞80岁时从原有42.5万减至20万个,PD患者少于10万个,出现症状时DA能神经元丢失50%以上,蓝斑、中缝核、迷走神经背核、苍白球、壳核、尾状核及丘脑底核等也可见轻度改变。
残留神经元胞浆中出现嗜酸性包涵体路易(Lewy)小体是本病重要病理特点,Lewy小体是细胞浆蛋白质组成的玻璃样团块,中央有致密核心,周围有细丝状晕圈。一个细胞有时可见多个大小不同的Lewy小体,见于约10%的残存细胞,黑质明显,苍白球、纹状体及蓝斑等亦可见,α-突触核蛋白和泛素是Lewy小体的重要组分。
3.神经生化改变
DA和乙酰胆碱(Ach)作为纹状体两种重要神经递质,功能相互拮抗,维持两者平衡对基底节环路活动起重要调节作用。脑内DA递质通路主要为黑质-纹状体系,黑质致密部DA能神经元自血流摄入左旋酪氨酸,在细胞内酪氨酸羟化酶(TH)作用下形成左旋多巴(L-dopa);再经多巴胺脱羧酶(DDC)作用生成多巴胺(DA);通过黑质-纹状体束,DA作用于壳核、尾状核突触后神经元,最后被分解成高香草酸(HVA)。
由于特发性帕金森病TH和DDC减少,使DA生成减少(左旋酪氨酸生成L-dopa减少,DA生成减少)。单胺氧化酶B(MAO-B)抑制可剂减少神经元内DA分解代谢,增加脑内DA含量。儿茶酚-氧位-甲基转移酶(COMT)抑制剂能减少L-dopa外周代谢,维持L-dopa稳定的血浆浓度。
PD患者黑质DA能神经元变性丢失、黑质-纹状体DA通路变性,纹状体DA含量显著降低(>80%),使Ach系统功能相对亢进,是导致肌张力增高、动作减少等运动症状的生化基础。近年发现中脑-边缘系统和中脑-皮质系统DA含量亦显著减少,可能导致智能减退、行为情感异常、言语错乱等高级神经活动障碍。DA递质减少程度与患者症状严重度一致,病变早期通过DA更新率增加(突触前代偿)和DA受体失神经后超敏现象(突触后代偿),临床症状可不明显(代偿期),随疾病进展出现典型PD症状(失代偿期)。基底节其他递质或神经肽,如去甲肾上腺素(NE)、5-羟色胺(5-HT)、P物质(SP)、脑啡肽(ENK)、生长抑素(SS)也有变化。
疾病预防:
帕金森预防
预防:迄今,帕金森病确切病因尚不十分清楚,因此,预防措施缺乏精确的针对性。但许多研究已证实,本文上述诸多危险因素与中脑黑质多巴胺神经元变性、坏死存在着一定的因果关系,若能针对危险因素采取相应的预防措施,对预防帕金森病的发病和延缓病程进展肯定是有益的。
一级预防(无病防病)
1、对有帕金森病家族史及有关基因携带者,有毒化学物品接触者,均应视为高危人群,须密切监护随访,定期体检,并加强健康教育,重视自我防护。
2、加大工农业生产环境保护的力度,减少有害气体、污水、污物的排放,对有害作业人员应加强劳动防护。
3、改善广大农村及城镇的饮水设施,保护水资源,减少河水、库水、塘水及井水的污染,保证广大人民群众能喝上安全卫生的饮用水。
4、老年人慎用吩噻嗪类、利血平类及丁酰苯类药物。
5、重视老年病(高血压、高血脂、高血糖、脑动脉硬化等)的防治,增强体质,延缓衰老,防止动脉粥样硬化,对预防帕金森病均能起到一定的积极作用。
二级预防(早发现、早诊断、早治疗)
1、早期诊断。帕金森病的亚临床期长,若能即早开展临床前期诊断技术,如嗅觉机能障碍、PET扫描、线粒体DNA、多巴胺抗体、脑脊液化学、电生理等检查,将亚临床期帕金森病尽早发现,采用神经保护剂(如维生素E、SOD、谷胱甘肽及谷胱甘肽过氧化物酶、神经营养因子、塞利吉林)治疗,可能会延缓整个临床期的过程。
2、帕金森病早期,虽然黑质和纹状体神经细胞减少,但多巴胺分泌却代偿性增加,此时脑内多巴胺含量并未明显减少,称代偿期,一般不主张用药物治疗,可采用理疗、医疗体育、太极拳、水疗、按摩、气功、针灸等治疗,以维持日常一般工作和生活,尽量推迟抗震颤麻痹药物应用的时间。但也有人主张早期应用小剂量左旋多巴以减少并发症,这要因人而异,择优选用。 3、帕金森病失代偿期应使用药物治疗。
三级预防(延缓病情发展、防止病残、改善生活质量)
1、积极进行非药物如理疗、体疗、针灸、按摩等及中西医药物或手术等综合治疗,以延缓病情发展。
2、重视心理疏导安抚和精神关爱,保证充足睡眠,避免情绪紧张激动,以减少肌震颤加重的诱发因素。
3、积极鼓励患者主动运动,如吃饭、穿衣、洗漱等。有语言障碍者,可对着镜子努力大声地练习发音。加强关节、肌力活动及劳作训练,尽可能保持肢体运动功能,注意防止摔跤及肢体畸形残废。
4、长期卧床者,应加强生活护理,注意清洁卫生,勤翻身拍背,防止坠积性肺炎及褥疮感染等并发症,帕金森病大部分死于肺部或其他系统如泌尿系统等的感染。注意饮食营养,必要时给予鼻饲,保持大小便通畅。以不断增强体质,提高免疫功能,降低死亡率。
疾病鉴别:
帕金森鉴别诊断
特发性PD须与家族性PD、Parkinson综合征鉴别,早期不典型病例须与遗传病或变性病伴Parkinson综合征鉴别。
1.家族性PD
约占10%,为不完全外显率常染色体显性遗传,可用DNA印迹技术、PCR和DNA序列分析等,检测α-突触核蛋白基因、Parkin基因突变,易感基因分析如细胞色素P450-2D6基因突变等。
2.Parkinson综合征 有明确病因,继发于药物、感染、中毒、脑卒中和外伤等。
(1)脑炎后Parkinson综合征:20世纪上半叶流行的昏睡性(von Economo)脑炎常遗留帕金森综合征,目前罕见。
(2)药物或中毒性Parkinson综合征:神经安定剂(酚噻嗪类及丁酰苯类)、利血平、胃复安、α-甲基多巴、锂、氟桂嗪等可导致帕金森综合征;MPTP、锰尘、CO、二硫化碳中毒或焊接烟尘亦可引起。
(3)动脉硬化性Parkinson综合征:多发性脑梗死偶导致Parkinson综合征,患者有高血压、动脉硬化及脑卒中史,假性球麻痹、病理征和神经影像学检查可提供证据。
(4)外伤性如拳击性脑病,其他如甲状腺功能减退、肝脑变性、脑瘤和正常压力性脑积水等可导致Parkinson综合征。
3.遗传病伴Parkinson综合征
(1)弥散性路易体病(diffuse Lewis body disease,DLBD):多见于60~80岁,痴呆、幻觉、帕金森综合征运动障碍为临床特征,痴呆早期出现,进展迅速,可有肌阵挛,左旋多巴反应不佳,但副作用极敏感。
(2)肝豆状核变性(Wilson病):可引起帕金森综合征,青少年发病,一或两侧上肢粗大震颤,肌强直、动作缓慢或不自主运动,肝损害和角膜K-F环,血清铜、铜蓝蛋白、铜氧化酶活性降低,尿铜增加等。
(3)亨廷顿(Huntington)病:运动障碍以肌强直、运动减少为主,易误诊为PD。
4.变性病伴Parkinson综合征
(1)多系统萎缩(MSA):累及基底节、脑桥、橄榄、小脑及自主神经系统,可有PD样症状,对左旋多巴不敏感。
包括:纹状体黑质变性(SND),表现运动迟缓、肌强直,可有锥体系、小脑和自主神经症状,震颤不明显。 Shy-Drager综合征(SDS),自主神经症状突出,直立性低血压、无汗、排尿障碍和阳萎,以及锥体束、下运动神经元和小脑体征等。橄榄脑桥小脑萎缩(OPCA),小脑及锥体系症状突出,MRI显示小脑和脑干萎缩。
(2)进行性核上性麻痹(PSP):可有运动迟缓和肌强直,早期姿势步态不稳和跌倒,垂直凝视不能,伴额颞痴呆、假性球麻痹、构音障碍及锥体束征,震颤不明显,左旋多巴反应差。
(3)皮质基底节变性(CBGD):表现肌强直、运动迟缓、姿势不稳、肌张力障碍和肌阵挛等,可有皮质复合感觉缺失、一侧肢体忽略、失用、失语和痴呆等皮质损害症状,眼球活动障碍和病理征,左旋多巴治疗无效。
(4)Alzheimer病伴Parkinson综合征。
(5)抑郁症:可有表情贫乏、言语单调、自主运动减少,PD患者常并存。抑郁症无肌强直和震颤,抗抑郁药试验治疗可能有助于鉴别。
(6)特发性震颤:多早年起病,姿势性或动作性震颤,影响头部引起点头或摇晃,PD典型影响面部、口唇。本病无肌强直和运动迟缓,约1/3的患者有家族史,饮酒或服心得安震颤明显减轻。
疾病检查:
帕金森检查
实验室检查
1.血清肾素活力降低、酪氨酸含量减少;黑质和纹状体内NE、5-HT含量减少,谷氨酸脱羧酶(GAD)活性较对照组降低50%。
2.CSF中GABA下降,CSF中DA和5-HT的代谢产物HVA含量明显减少。
3.生化检测 放免法检测CSF生长抑素含量降低。尿中DA及其代谢产物3-甲氧酪胺、5-HT和肾上腺素、NE也减少。
影像学检查
1.CT、MRI影像表现
由于帕金森病是一种中枢神经系统退性变疾病,病理变化主要在黑质、纹状体、苍白球、尾状核以及大脑皮质等处,所以,CT影像表现,除具有普遍性脑萎缩外,有时可见基底节钙化。MRI除能显示脑室扩大等脑萎缩表现外,T2加权像在基底节区和脑白质内常有多发高信号斑点存在。
2.SPECT影像表现
(1)通过多巴胺受体(DAR)的功能影像:多巴胺受体广泛分布于中枢神经系统中多巴胺能通路上,其中主要是黑质、纹状体系统,DAR(DL)分布于纹状体非胆碱能中间神经元的胞体;DAR(D2)位于黑质、纹状体多巴胺能神经元胞体。
SPECT是将放射性核素,目前主要是123I-IBZM,131I-IBZM,特异性D2受体标记物,静脉注入人体后,通过在基底节区域的放射活性与额叶、枕叶或小脑放射活性的比值,反映DAR受体数目和功能,来诊断早期帕金森病。如果早期采用多巴制剂治疗患者,起病对侧脑DAR(D2)上调。长期服用多巴制剂的中晚期帕金森病患者,脑中基底节/枕叶和基底节/额叶比值减少,SPECT功能影像只能检测DAR受体数目,不能帮助确诊是否为原发性帕金森病,但是可以区别某些继发性帕金森病,还可用作帕金森病病性演变和药物治疗效果指标。
(2)通过多巴胺转运蛋白(DAT)功能显像:多巴胺转运蛋白(DAT)如何转运多巴胺(DA)尚不清楚,DAT主要分布于基底节和丘脑,其次为额叶。DAT含量与帕金森病的严重程度是存在着正相关性,基底节DAT减少,在早期帕金森病患者表现很显著。
SPECT采用11C-WIN35428、123Ibeta;-CIT,通过静脉注入人体后,检测基底节/小脑活性比值以及丘脑/小脑活性比值,反映中枢不同区域DAT数量。早期帕金森病患者,基底节区域DAT数目明显减少。
3.PET功能影像
正电子发射断层扫描(PET)诊断帕金森病,其工作原理和方法与SPECT基本相似,目前主要是依赖脑葡萄糖代谢显像,一般采用18F脱氧葡萄糖(18FDG)。因为在帕金森病病人早期,纹状体局部葡萄糖代谢率就中度降低,晚期葡萄糖代谢率进一步降低。用PET的受体显像剂很多,PET神经递质功能显像剂主要是用18F-多巴-PET(18FD-PET)等核素,基本原理同SPECT。PET可对帕金森病进行早期诊断,可作帕金森病高危人群中早期诊断,是判断病情严重程度的一种客观指标。
疾病就诊:
1、描述就诊原因(从什么时候开始,有什么不舒服?)
2、是否到过医院就诊,做过那些检查,检查结果是什么?
3、不适的感觉是否由明显的因素引起?
4、治疗情况如何?
5、有无药物过敏史?
疾病治疗:
帕金森一般治疗
帕金森西医治疗
1.PD早期治疗
PD早期黑质-纹状体系统存留的DA神经元可代偿地增加DA合成,推荐采用理疗(按摩、水疗)和体育疗法(关节活动、步行、平衡及语言锻炼、面部表情肌操练)等,争取患者家属配合,鼓励患者多主动运动,尽量推迟药物治疗时间。若疾病影响患者日常生活和工作,需药物治疗。
2.药物治疗
PD目前仍以药物治疗为主,恢复纹状体DA与Ach递质系统平衡,应用抗胆碱能和改善DA递质功能药物,改善症状,不能阻止病情发展。
用药原则:
①从小剂量开始,缓慢递增,尽量用较小剂量取得满意疗效;
②治疗方案个体化,根据患者年龄、症状类型和程度、就业情况、药物价格和经济承受能力等选择药物;
③不应盲目加用药物,不宜突然停药,需终生服用;
④PD药物治疗复杂,近年来推出的辅助药物DR激动药、MAO-B抑制剂、儿茶酚-氧位-甲基转移酶(COMT)等,与复方多巴合用可增强疗效、减轻症状波动、降低复方多巴剂量,单独使用疗效不理想,应权衡利弊,适当选择联合用药。
(1)抗胆碱能药:
对震颤和强直有效,对运动迟缓疗效较差,适于震颤明显年龄较轻患者。常用安坦(artane)1~2mg口服,3次/d;开马君(kemadrin)2.5mg口服,3次/d,逐渐增至20~30mg/d。其他如苯甲托品(cogentin)、环戊丙醇(cycrimine)、安克痉(akineton)等,作用与安坦相似。副作用包括口干、视物模糊、便秘和排尿困难,严重者有幻觉、妄想。青光眼及前列腺肥大患者禁用,可影响记忆功能,老年患者慎用。
(2)金刚烷胺(amantadine):
促进DA在神经末梢释放,阻止再摄取,并有抗胆碱能作用,是谷氨酸拮抗药,可能有神经保护作用,可轻度改善少动、强直和震颤等,早期可单独或与安坦合用。起始剂量50mg,2~3次/d,1周后增至100mg,2~3次/d,一般不超过300mg/d,老年人不超过200mg/d。药效可维持数月至1年。副作用较少,如不安、意识模糊、下肢网状青斑、踝部水肿和心律失常等,肾功能不全、癫痫、严重胃溃疡和肝病患者慎用,哺乳期妇女禁用。也可用其衍生物盐酸美金刚烷(memantine hydrochloride)。
(3)左旋多巴(L-dopa)及复方左旋多巴:
L-dopa是治疗PD有效药物或金指标。作为DA前体可透过血脑屏障,被脑DA能神经元摄取后脱羧变为DA,改善症状,对运动减少有特殊疗效。由于95%以上的L-dopa在外周脱羧成为DA,仅约1%通过BBB进入脑内,为减少外周副作用,增强疗效,多用L-dopa与外周多巴脱羧酶抑制剂(DCI)按4∶1制成的复方制剂(复方L-dopa),用量较L-dopa减少3/4。
复方L-dopa剂型:包括标准片、控释片、水溶片等。标准片如美多巴(madopar)和帕金宁(sinemet):
①Madopar由L-dopa与苄丝肼按4∶1组成,美多巴250为L-dopa 200mg 苄丝肼50mg,美多巴125为L-dopa100mg 苄丝肼25mg;国产多巴丝肼胶囊成分与美多巴相同;
②帕金宁(Sinemet 250和Sinemet 125)由L-dopa与卡别多巴按4∶1组成。
控释剂包括两种:
①息宁控释片(sinemet CR):L-dopa 200mg 卡别多巴50mg,制剂中加用单层分子基质结构,药物不断溶释,达到缓释效果,口服后120~150min达到血浆峰值浓度;片中间有刻痕,可分为半片服用,保持缓释特性;
②美多巴液体动力平衡系统(Madopar-HBS):L-dopa 100mg 苄丝肼25mg及特殊赋形剂组成,胶囊溶解时药物基质表面形成水化层,通过弥散作用逐渐释放。
水溶片有弥散型美多巴(madopar dispersible),剂量为125mg,由L-dopa 100mg 苄丝肼25mg组成。其特点易在水中溶解,便于口服,吸收迅速,很快达到治疗阈值浓度,使处于关闭状态的PD患者在短时间内(10min左右)迅速改善症状,且作用维持时间与标准片基本相同。该剂型适用于有吞咽障碍或置鼻饲管、清晨运动不能开期延迟、下午关期延长、剂末肌张力障碍的PD患者。
用药时机:何时开始复方L-dopa治疗尚有争议,长期用药会产生疗效减退、症状波动及运动障碍等并发症。一般应根据患者年龄、工作性质、疾病类型等决定用药。年轻患者可适当推迟使用,早期尽量用其他抗PD药,患者因职业要求不得不用L-dopa时应与其他药物合用,减少复方L-dopa剂量。年老患者可早期选用L-dopa,因发生运动并发症机会相对较少,对合并用药耐受性差。
用药方法:从小剂量开始,根据病情逐渐增量,用最低有效量维持。
①标准片:复方L-dopa开始用62.5mg(1/4片),2~3次/d,根据需要逐渐增至125mg,3~4次/d;最大剂量不超过250mg,3~4次/d;空腹(餐前1h或餐后2h)用药疗效好;
②控释片:优点是减少服药次数,有效血药浓度稳定,作用时间长,可控制症状波动;缺点是生物利用度较低,起效缓慢,标准片转换成为控释片时每天剂量应相应增加并提前服用;适于伴症状波动或早期轻症患者;
③水溶片:易在水中溶解,吸收迅速,10min起效,作用维持时间与标准片相同,适于吞咽障碍、清晨运动不能、“开关”现象和剂末肌张力障碍患者。
副作用:周围性副作用常见恶心、呕吐、低血压和心律失常(偶见)等,用药后可逐渐适应,餐后服药、加用吗叮啉可减轻消化道症状。中枢性副作用包括症状波动、运动障碍和精神症状等,症状波动和运动障碍是常见的远期并发症,多在用药4~5年后出现。闭角型青光眼、精神病患者禁用。
(4)DA受体激动药:
DA包括五种类型受体,D1R和D2R亚型与PD治疗关系密切。DR激动药共同作用特点是:
①直接刺激纹状体突触后DR,不依赖于DDC将L-dopa转化为DA发挥效应;
②血浆半衰期(较复方多巴)长;
③可能对黑质DA能神经元有保护作用。早期DR激动药与复方多巴合用,不仅能提高疗效,减少复方多巴用量,且可减少或避免症状波动或运动障碍发生。
适应证:PD后期患者用复方多巴治疗产生症状波动或运动障碍,加用DR激动药可减轻或消除症状,减少复方多巴用量。疾病后期因黑质纹状体DA能系统缺乏DDC,不能把外源性L-dopa脱羧转化为DA,用复方多巴完全无效,用DR激动药可能有效。单用DA受体激动药疗效不佳,一般主张与复方L-dopa合用,发病年龄轻的早期患者可单独应用。应从小剂量开始,渐增量至获得满意疗效而不出现副作用。副作用与复方L-dopa相似,症状波动和运动障碍发生率低,体位性低血压和精神症状发生率较高。
常用制剂:主要是溴隐亭、培高利特。
①溴隐亭(bromocriptine):激活D2受体,开始0.625mg/d,每隔3~5天增加0.625mg,通常治疗剂量7.5~15mg/d,分3次服;副作用与左旋多巴类似,错觉和幻觉常见,精神病史患者禁用,相对禁忌证包括近期心肌梗死、严重周围血管病和活动性消化性溃疡等;
②培高利特(pergolide):激活D1和D2两类受体,开始0.025mg/d,每隔5天增加0.025mg,一般有效剂量0.375~1.5mg/d,最大不超过2.0mg/d,1~3h达血浆峰值浓度,半衰期较长(平均30h),较溴隐亭抗PD作用稍强,作用时间亦长,溴隐亭治疗无效时改用培高利特可能有效;
③泰舒达缓释片(trastal SR):化学成分为吡贝地尔,是选择性D2/D3多巴胺受体激动药,剂量为150~250mg/d,对中脑-皮质和边缘叶通路D3R有激动效应,改善震颤作用明显,对强直和少动也有作用;
④麦角乙脲(lisuride):具有较强选择性D2R激动作用,对D1R作用很弱,从小剂量开始,0.05~0.1mg/d,逐渐增量,平均有效剂量为2.4~4.8mg/d;按作用-剂量比,作用较溴隐亭强10~20倍,半衰期短(平均2.2h),作用时间短,为水溶性,可静脉或皮下输注泵应用,用于复方多巴治疗出现明显“开-关”现象;
⑤阿朴吗啡(apomorphine):D1和D2R激动药,可显著减少“关期”状态,对症状波动,尤其开-关现象和肌张力障碍有明显疗效,采取笔式注射法给药后5~15min起效,有效作用时间60min,每次给药0.5~2mg,每天可用多次,便携式微泵皮下持续灌注法可使患者每天保持良好运动功能;也可经鼻腔给药,但长期用药可刺激鼻黏膜;
⑥卡麦角林(cabaser):是所有DR激动药中半衰期最长(70h),作用时间最长,适于PD后期长期应用复方多巴产生症状波动和运动障碍患者,有效剂量2~10mg/d,平均4mg/d,只需1次/d,较方便;
⑦普拉克索(Pramipexole,0.125mg,3次/d,逐渐加量至0.5~1.0mg,3次/d)和罗吡尼洛(Ropinirole,0.25mg,3次/d,逐渐加量至2~4mg,3次/d),均非麦角衍生物,无麦角副作用,用于早期或进展期PD,症状波动和运动障碍发生率低,常见意识模糊、幻觉及直立性低血压。
(5)单胺氧化酶B(MAO-B)抑制剂:
抑制神经元内DA分解,增加脑内DA含量。合用复方L-dopa有协同作用,减少L-dopa约1/4用量,延缓开关现象,有神经保护作用。常用思吉宁(selegiline)即丙炔苯丙胺(deprenyl)2.5~5mg,2次/d,宜早、午服用,傍晚服用可引起失眠。副作用有口干、胃纳少和体位性低血压等,胃溃疡患者慎用。Lazabemide(Ro19-6327)亦系MAO-B抑制剂,目前临床应用报道不多。
有学者主张此类药与维生素E合用,称DATA-TOP方案(deprenyl and tocopherol antioxidation therapy of Parkinsonism),作为神经保护剂用于早期轻症患者,可能延缓疾病进展。维生素E是天然自由基清除剂,有抗氧化作用,PD早期尤其未经治疗患者用维生素E与丙炔苯丙胺可能减缓黑质细胞变性、延缓疾病进展。近年国外有人提倡丙炔苯丙胺2.5mg口服,1次/d,渐加至2.5mg,2次/d,再加至5mg,2次/d;同时服用维生素E 2000U,1次/d。但目前对此方案仍有争议,须继续观察评价。
(6)儿茶酚-氧位-甲基转移酶(COMT)抑制剂:
抑制L-dopa外周代谢,维持L-dopa稳定血浆浓度,加速通过BBB,阻止脑胶质细胞内DA降解,增加脑内DA含量。与美多巴或息宁合用增强后者疗效,减少症状波动反应,单独使用无效。副作用可有腹泻、头痛、多汗、口干、转氨酶升高、腹痛、尿色变浅等,用药期间须监测肝功能。
常用制剂:
①托可朋(tolcapone):亦名答是美(tasmar),100~200mg口服,3次/d,副作用有腹泻、意识模糊、运动障碍和转氨酶升高等,应注意肝脏毒副作用;具有周围和中枢COMT抑制作用,临床试验显示,应用复方多巴疗效减退的69例PD加用托可朋100~150mg,3次/d,疗程6个月,有效率98.5%,无明显毒副作用,可与复方多巴和MAO-B抑制剂合用;
②恩他卡朋(entacapone):亦名珂丹(comtan),是周围COMT抑制剂,100~200mg口服,5次/d为宜,与托可朋不同的是迄今无严重肝功能损害报道。
(7)兴奋性氨基酸(EAA)受体拮抗药及释放抑制剂:
EAA可损害黑质细胞,抑制剂有神经保护作用,可增强L-dopa作用。但目前尚无临床有效治疗的报道。
(8)铁螯合剂:
PD患者黑质Fe2 浓度明显增加,铁蛋白含量显著减少。给予铁螯合剂可降低Fe2 浓度,减少氧化反应。目前常用21-氨基类固醇(21-aminosteroide),可通过血脑屏障与Fe2 结合,抑制脂质过氧化,对黑质细胞有保护效应。
(9)神经营养因子(neurotrophic factors):
对神经元发育、分化及存活起重要作用,选择性作用于DA能神经元的神经营养因子有助于PD防治。神经营养因子包括酸性及碱性成纤维细胞生长因子(aFGF、bFGF)、上皮生长因子(EGF)、睫状神经营养因子(CNTF)、脑源性神经营养因子(BDNF)、胶质细胞源性神经营养因子(GDNF)及Neurturin等。GDNF和Neurturin对中脑DA能神经元特异性强。
(10)中药或针灸对PD治疗有一定的辅佐作用,需与西药合用,单用疗效不理想。
3.外科治疗
立体定向手术治疗PD始于20世纪40年代,近年来随着微电极引导定向技术的发展,利用微电极记录和分析细胞放电特征,可精确定位引起震颤和肌强直的神经元,达到细胞功能定位,可显著提高手术疗效和安全性。手术可纠正基底节过高的抑制性输出,适应证为药物治疗失效、不能耐受或出现运动障碍(异动症)的患者,年龄较轻,症状以震颤、强直为主且偏于一侧者效果较好,术后仍需用药物治疗。
(1)苍白球毁损术(pallidotomy):
近年来随着微电极引导定向技术的发展,使定位精确度达到0.1mm,进入到细胞水平,达到准确功能定位,确定电极与苍白球各结构及相邻视束和内囊的关系,有助于寻找引起震颤和肌张力增高的神经元。用此法确定靶点,手术效果较好,改善PD运动症状,尤其运动迟缓,很少产生视觉受损等并发症。
(2)丘脑毁损术:
是用立体定向手术破坏一侧丘脑腹外侧核、豆状襻及丘脑底核,对PD的震颤疗效较好,最佳适应证是单侧严重震颤。单侧丘脑毁损术并发症较少,双侧毁损术可引起言语障碍、吞咽困难及精神障碍等并发症,不主张采用。
(3)深部脑刺激疗法(deep brain stimulation,DBS):
是将高频微电极刺激装置植入PD患者手术靶点,高频电刺激产生的电压和频率高于病变神经元产生的电压和频率,从而起到抑制作用。DBS优点是定位准确、损伤范围小、并发症少、安全性高和疗效持久等,缺点是费用昂贵。美国FDA已批准临床应用DBS治疗PD。
(4)立体定向放射治疗(γ-刀,X-刀):
利用立体定向原理,用计算机精确计算靶点,一次大剂量窄束高能射线精确地聚焦破坏靶点,靶点外正常组织受剂量极小。射线包括60钴(60CO)产生的γ-射线(γ-刀)及直线加速器产生的X射线(X-刀)。适应证与立体定向毁损术相同,但疗效不如后者,副作用较多,目前不推荐使用。
4.细胞移植及基因治疗
细胞移植是将自体肾上腺髓质或异体胚胎中脑黑质细胞移植到患者纹状体,纠正DA递质缺乏,改善PD运动症状。酪氨酸羟化酶(TH)和神经营养因子基因治疗正在探索中,是有前景的新疗法。将外源TH基因通过exvivo或invivo途径导入动物或患者脑内,导入的基因经转录、翻译合成TH,促使形成DA。目前存在供体来源困难、远期疗效不肯定及免疫排斥等问题。
5.康复治疗
对患者进行语言、进食、行走及各种日常生活训练和指导,对改善生活质量十分重要。晚期卧床者应加强护理,减少并发症发生。康复包括语音语调训练,面肌锻炼,手部、四肢及躯干锻炼,松弛呼吸肌锻炼,步态及平衡锻炼,姿势恢复锻炼等。
疾病护理:
帕金森一般护理
帕金森护理
一、预后
PD是慢性进展性疾病,目前无根治方法,多数患者发病数年仍能继续工作,也可迅速发展致残。疾病晚期可因严重肌强直和全身僵硬,终至卧床不起。死因常为肺炎、骨折等并发症。
二、认清帕金森病的五大误区:
误区之一:吸烟能防治帕金森
吸烟有害健康(健康食品),妇孺皆知,但社会上曾流传吸烟可以防治帕金森病之说。事实上,这种民间说法并非完全空穴来风,现在的研究表明烟草中的主要成分尼古丁以及4-苯吡啶和肼可以抑制导致帕金森病的某些神经毒素的毒性作用,促进其降解,提高脑内神经营养因子的水平,从而保护脑内多巴胺能神经元。但这决不意味着吸烟就对帕金森病有百利而无一害,因为同时的大量调查研究显示,除了可以导致肺癌、肺气肿等疾病的发病率增高外,吸烟对于已经确诊的帕金森病患者并无明显的保护作用,并且长期大量的吸烟可导致脑动脉硬化,反而会增加罹患帕金森病乃至老年性痴呆等其他严重疾病的机会;此外,在日常生活中,许多人都有饮绿茶和喝咖啡的习惯,调查显示长期饮用绿茶和咖啡可以降低帕金森病的发病率。目前的研究发现绿茶中的主要成分茶多酚和咖啡的主要成分咖啡因可以增加脑内多巴胺的含量、抑制神经毒素的作用,从而对多巴胺能神经元具有保护作用,所以长期饮用的人不易患上帕金森病。因此,保持良好的生活方式对于帕金森病的预防是大有裨益的。
误区之二:保健(保健食品)品当成灵丹妙药;
常常有患者会被一些医疗广告所诱导,误以为帕金森病用某些偏方可以根治而贸然相信服用,既耽误了病情,又加重了经济负担,往往得不偿失。目前很多人深信注射神经节苷脂对治疗帕金森有效果,但事实是否如此?有些动物试验表示,神经节苷脂可以协助旁支神经萌芽,从而帮助康复。于是出现了神经节苷脂对脑缺血有保护作用的说法。但人身上的临床试验结果其实并不支持这种想法。截至目前尚无正式的科学报告出来,它作为治疗帕金森病的药物也还没有通过临床验证!直至今年,已经公开发表了的12个随机对照试验,参加人数达2265人。大部分试验质素欠佳,影响可靠程度。缺点包括随机分组方法含糊、没有治疗意向分析、统计数字交待不清楚等等。
除了脑梗塞之外,也有人用神经节苷脂来医脊髓创伤和脑创伤。同样地,这种治疗法目前也缺乏科学证据支持。脊髓创伤方面,最初试验表示可能有用,但当时是和皮质激素并用,因此疗效难以确定。之后进行单独使用神经节苷脂的试验已完成一段时期,详细结果却迟迟未见正式发表。但据研究者所透露,在用药六月后,试验组和对照组似乎无分别。至于用来治疗脑创伤,更至今仍无严格的科学试验。
误区之三:轻信这刀那刀能根治
帕金森病的治疗通常不是一种方法所能解决的,大多数患者一般以药物治疗为主,配合康复治疗和中医药治疗;符合手术指征的难治性帕金森病患者还需进行外科治疗,但手术后仍然要服药和辅以其他治疗。
在我国的一些医院,曾经引进各种设备如gamma刀、ldquo细胞刀;等来治疗帕金森病,经过一段时间的临床效果观察,发现这些治疗手段的效果并不如想象的那么理想,有的只能改善部分症状,有的甚至会引起患者失明等严重的副作用。据专家介绍,目前欧美等发达国家已经基本放弃了这些治疗方法。
据介绍,目前对帕金森病治疗,公认的是以左旋多巴特别是复方左旋多巴为金标准的药物治疗,另外就是被誉为近年来帕金森病外科治疗中的重大突破,目前已被医学界公认为帕金森病的有效治疗方案的脑深部电极刺激术(即俗称脑起搏器)。而国外近年来研发出经颅磁刺激新型治疗方法,则可以说是填补了帕金森疾病物理疗法的空白。
误区之四:误当颈病腰病治
刚刚发病的帕金森病患者往往会感觉肢体无力,肢端发硬发僵不灵活,有时还会有酸困疼痛的感觉,病人往往以为是颈椎或腰椎出了问题,到骨科拍片、做CT检查;医生对疾病早期单侧肢体出现症状的患者也易误诊其为颈椎、腰椎疾病,甚至有些腿部起病的帕金森病患者还被误做了椎间盘手术。其实两种病的疼痛感觉是完全不一样的,必要时请神经内、外科会诊,尽量避免误诊。
误区之五:寄望脑移植手术
目前有些医院打出所谓的脑移植手术招牌,其实质只是用胚胎脑细胞悬液采用定向手术进行注射,通过局部小毁损来减轻症状,而一旦占位效应解除,症状又会重新出现。还有干细胞脑移植术,也还在实验阶段,虽有细胞成活,也还不能取代原细胞发挥作用。2003年美国FDA批准一项实验计划,该方法是向脑内注射一种能产生多巴胺的转基因病毒,是一种新思路,尚在临床实验中。由此可见,寻找一种能彻底根治帕金森病的方法还有待人类进一步探索。
疾病饮食:
帕金森饮食原则
帕金森饮食保健
一、帕金森病食疗方(下面资料仅供参考,详细需要咨询医生)
1、枣仁龙眼汤:龙眼肉、炒枣仁各15克。将龙眼肉、炒枣仁加入水煎成汁,再加适量白蜜即成。每日2次,早、晚服用。对久患帕金森氏病、气血亏虚者有补益作用。
2、沙棘菊花饮:沙棘50克,菊花10克。将沙棘、菊花洗净后共同煎汤,每日2次,可早、晚服用一次,也可代茶饮。适用于帕金森氏病合并高脂血症。
3、陈皮砂仁酸枣粥:陈皮5克,砂仁10克,酸枣15克,粳米适量。将砂仁先煮成汤,再放入粳米,酸枣煮成粥后,再放入陈皮,稍混后即可食用。每日2次,早、晚服食。具有镇静作用。
4、天麻炖鹌鹑
材 料:鹌鹑一只,天麻15克。
制作方法:鹌鹑去毛及内脏,将天麻填入其肚内,用线捆住,用水炖熟,加食盐、味精。
食用方法:去天麻,吃肉喝汤,隔日一次。
5、天麻炖猪脑
材 料:天麻10克,猪脑花100克。
制作方法:将上述材料放入沙锅内,加水适量,以文火炖一小时左右。
服用方法:调味后喝汤食猪脑花,每日服用一次,或隔日服用一次。
6、枸杞蒸羊脑
材 料:枸杞子50克,羊脑花一具
制作方法:将上述材料放入容器,加水适量,加姜末、葱节、料酒、食盐、隔水蒸熟后即可食用。
服用方法:每日分两次吃。
7、天麻鱼头汤
材 料:天麻15克,川芎10克,鲜鲤鱼头1个。
制作方法:将天麻、川芎泡软后切薄片放入鱼头中,置盘内,加葱姜,再加适量清水上笼约30分钟。
食用方法:食鱼肉喝汤,隔日一次。
8、枸杞血藤饮
材 料:枸杞子20克,鸡血藤15克,红花5克。
制作方法:取上述材料,加水500毫升,煎至300毫升,将药液倒入碗中,放黄酒30克。
服用方法:早晚分两次饮服,每日一剂。
9、核桃黄酒泥
材 料:核桃仁15个,白糖50克。
制作方法:将上述材料放在砂罐或瓷碗中,用擀面杖捣成泥状,再放入锅中,加黄酒50毫升,用小火煎煮10分钟。
服用方法:每日食用两次。
二、帕金森病吃哪些对身体好?
(1) 普食:和正常人的饮食基本相同,适用于咀嚼能力尚好的帕金森病病人。
(2) 软食:适用于咀嚼能力和消化能力减低的病人,可采用易消化(消化食品)、易咀嚼、细软、无刺激 的食品。
(3) 半流质软食:适用于咀嚼、吞咽功能受一定限制的病人,可选用面片、稀酸、豆腐脑、 蛋羹、鸡蛋汤等。
(4) 流质:适用于晚期病人,咀嚼、吞咽功能明显障碍者。如能由口腔进食者要尽量由口腔 进食,缓慢以汤匙或奶瓶喂食,防止呛咳。严重病人 必要时给予鼻饲。一般选用牛奶、豆浆 、米汁、麦乳精、藕粉、肉汤、菜汁等作为鼻饲饮食。
(5) 此外,帕金森病患者便秘是很常见的。饮食中给予适量的新鲜蔬菜(蔬菜食品)、水果(水果食品)和蜂蜜(蜂蜜食品)很为必要, 既能缓解便秘(便秘食品),又能补充维生素(维生素食品)类。要避免刺激性食物及烟酒等。
(6)多吃谷类和蔬菜瓜果。从谷类中主要能得到碳水化合物、蛋白质、膳食纤维和维生素B 等营养,并能获取身体所需的能量。碳水化合物通常不影响左旋多巴的药效。
三、帕金森病最好不要吃哪些食物?
1、限制蛋白质(蛋白质食品)。帕金森氏病患者的热能摄入以维持正常体重为宜。过度消瘦与肥胖均不利于治疗。服用多巴胺治疗者宜限制蛋白质摄入量。因为蛋白质可影响多巴胺的治疗效果。蛋白质摄入量限制在每日每公斤体重0.8克以下,全日总量约40克~50克。在限制范围内多选用乳、蛋、肉、豆制品等优质蛋白质。
2、禁烟酒及刺激性食品,如咖啡、辣椒、芥末、咖喱等。
3、不吃肥肉、荤油和动物内脏,有助于防止由于饱和脂肪和胆固醇摄入过多给身体带来的不良影响。饮食中过高的脂肪也会延迟左旋多巴药物的吸收,影响药效。





